Análise genética de casos de ALS na população isolada da ilha de Malta
European Journal of Human Genetics volume 29 , Páginas604–614 ( 2021 )
Abstrato
Isolados genéticos são ferramentas atraentes para mapear genes de doenças hereditárias. O arquipélago de Malta, um microestado soberano no sul da Europa, é o lar de uma população geográfica e culturalmente isolada. Aqui, investigamos a epidemiologia e o perfil genético de pacientes malteses com esclerose lateral amiotrófica (ELA), identificados ao longo de uma janela de 2 anos. Os casos eram predominantemente do sexo masculino (66,7%), com início predominante dos sintomas na medula espinhal (70,8%). O início da doença ocorreu por volta da meia-idade (idade mediana: 64 anos, homens; 59,5 anos, mulheres); 12,5% tinham ALS familiar (fALS). A taxa de incidência anual foi de 2,48 (IC de 95% 1,59–3,68) por 100.000 pessoas-ano. A taxa de incidência entre homens e mulheres foi de 1,93: 1. Prevalência foi de 3,44 (IC de 95% 2,01-5,52) casos por 100.000 habitantes em 31 de rDezembro de 2018. O sequenciamento do genoma completo nos permitiu determinar variantes raras de DNA que alteram a sequência codificadora de proteínas de genes associados a ALS. Curiosamente, a coorte de pacientes com ALS em Malta foi considerada negativa para variantes deletérias nos genes C9orf72 , SOD1 , TARDBP ou FUS , que são os genes ALS mais comumente mutados em todo o mundo. No entanto, expansões de repetição associadas a ALS foram identificadas em ATXN2 e NIPA1 . Variantes previstas para serem prejudiciais também foram detectadas em ALS2 , DAO , DCTN1 , ERBB4 , SETX , SCFD1 e SPG11. Um total de 40% dos pacientes com ELA esporádica tinham uma variante rara e deletéria ou expansão repetida em um gene associado a ELA, enquanto a causa genética de dois terços dos casos de FALS não pôde ser identificada para genes de ELA conhecidos ou loci de risco. Isso garante mais estudos para elucidar novos genes que causam ALS neste isolado de população única.
Introdução
A esclerose lateral amiotrófica (ELA) é uma doença neurodegenerativa com início na idade adulta e progressão rápida. O início é tipicamente acompanhado por sinais clínicos de degeneração do neurônio motor superior e / ou inferior e os pacientes geralmente apresentam fraqueza nos músculos bulbares, apenas nos membros ou em ambas as regiões simultaneamente. Cerca de 15-20% das pessoas com ALS experimentam declínio cognitivo progressivo, levando à demência. A condição conhecida como demência frontotemporal resulta da degeneração dos lobos frontal e temporal [ 1 , 2 ]. A incidência de ALS em populações europeias é de dois a três casos por ano por 100.000, enquanto a prevalência pode chegar a 10 / 100.000 [ 3] A ELA é classificada como familiar (fALS) na presença de uma história familiar clara da doença e esporádica (sALS) quando esta está ausente. A taxa de fALS entre os registros de base populacional em perspectiva é de cerca de 5% [ 4 ]. No entanto, estudos em gêmeos e coortes de caso-controle europeias descobriram que a herdabilidade de ALS se aproxima de 40% [ 5 , 6 , 7 ]. Até o momento, variantes em qualquer um de mais de 40 genes foram relatados como causadores de fALS monogênicos com mais da metade dos casos explicados por variantes causais altamente penetrantes residindo em C9orf72 (23%), SOD1 (19%), TARDBP (3%) ou FUS (3%) [ 8 ]. Variantes nesses genes [8 , 9 , 10 ] e loci de risco genético que incluem ATXN2 [ 11 ], UNC13A [ 12 ], SARM1 [ 13 ], C21orf2 , SCFD1 e MOBP [ 13 ] também foram expostos em casos de sALS. Estima-se que as variantes genéticas contribuam com 14 a 17% dos pacientes europeus com sALS ou aqueles com ascendência europeia [ 9 , 10 ]. Tudo isso ressalta a contribuição substancial dos fatores genéticos para a etiologia da doença de ALS.
As descobertas genéticas geralmente levam a novos insights sobre os mecanismos moleculares da ELA. Por exemplo, o agrupamento de genes de acordo com suas funções fisiológicas conhecidas identificou a ribostase, a proteostase e a dinâmica do citoesqueleto como as principais vias celulares envolvidas na patogênese da ELA [ 14 ]. É importante ressaltar que a descoberta de genes está levando ao desenvolvimento de tratamentos específicos para genótipos [ 15 ]. Fatores genéticos adicionais ainda precisam ser encontrados para ALS. O uso de populações geograficamente e / ou culturalmente isoladas para mapear novos genes ALS é uma estratégia que permanece relativamente inexplorada, apesar dos benefícios que incluem efeitos fundadores, diversidade genética reduzida e heterogeneidade ambiental mínima [ 16] Isso nos estimulou a estudar a população nativa de Malta, um microestado soberano no meio do Mar Mediterrâneo. Consistindo de um arquipélago de três ilhas habitadas (área total de 316 km 2 ), a população de Malta atualmente é de cerca de 514.564, com base nos dados do Escritório Nacional de Estatísticas de Malta (NSO) em 2019. Eventos de semeadura populacional ocorreram há mais de 7.000 anos por colonos vindos de vizinhos Sicília e com base em análises de DNA mitocondrial, cromossomo Y e marcadores de DNA autossômico, as influências dos colonizadores nos séculos que se seguiram foram mínimas [ 17 , 18 , 19] Os malteses são a única população europeia que fala uma língua semítica, sublinhando ainda mais o seu relativo isolamento de outras comunidades que habitam a Europa. Os sucessos anteriores no mapeamento genético de doenças raras na população de Malta são encorajadores [ 20 , 21 ]. Aqui, primeiro investigamos a incidência e prevalência de ALS nas ilhas maltesas, em um período de 2 anos. Em segundo lugar, realizamos uma pesquisa genética inicial relatando variantes raras em regiões codificadoras de proteínas e locais de splice de consenso de todos os genes ALS monogênicos atualmente conhecidos e loci de risco genético em pacientes malteses com ALS em comparação com controles pareados.
materiais e métodos
Participantes
A vigilância e o recrutamento dos participantes ocorreram ao longo de uma janela de 2 anos, de 2017 a 2018. Pacientes com diagnóstico de ELA provável ou definitiva, encaminhados pela associação nacional de Doença do Neurônio Motor, neurologistas consultores, clínicos gerais e unidades de neurofisiologia foram convidados a participar de nosso estudo . Alternativamente, os pacientes ou seus parentes fizeram contato direto com nosso laboratório manifestando a vontade de participar do estudo. Pacientes participantes preencheram os critérios revisados de El Escorial para ALS [ 22 , 23] Os pacientes com fALS foram identificados como tendo uma história familiar de ELA autorreferida, ou provável ELA, definida como a presença de pelo menos um parente de primeiro grau. No total, 24 pacientes foram incluídos neste estudo. A amostragem de sangue foi excluída para um caso de sALS devido à condição de deterioração do paciente. Os membros da família afetados para todos os casos de fALS faleceram, impedindo-nos de amostrá-los. Os controles, que foram verificados em uma proporção de caso-controle de aproximadamente 2: 1, combinaram os pacientes por idade, sexo e região geográfica. A aprovação ética para a coleta de amostras, desenho do estudo e criação do Registro e Biobanco Malta ALS / MND foi concedida pelo Comitê de Ética em Pesquisa da Universidade de Malta. O consentimento informado por escrito para participar foi obtido de todos os pacientes e / ou familiares, bem como dos controles.
Informação fenotípica
As informações fenotípicas foram coletadas por meio de um questionário detalhado, além de um exame clínico. Cada amostra é, portanto, acompanhada por um conjunto de dados principal que inclui idade, sexo, ocupação, local de início, data de início da doença, história familiar, pontuação da ALS Functional Rating Scale revisada (ALSFRS-R), tônus muscular para membros superiores e inferiores , potência muscular graduada de acordo com a escala MRC e estado dos reflexos. Informações sobre possíveis fatores de risco ambientais, incluindo atividade física, tabagismo e consumo de álcool, também foram coletadas. Um ensaio bioquímico para testar os níveis de creatina quinase (CK) no recrutamento também foi utilizado.
Cálculos de incidência e prevalência
O denominador para o cálculo da taxa de incidentes foi a soma da população total de Malta em 2017 e 2018. Durante o período do estudo, a população de Malta aumentou de 475.701 para 493.559. Taxas de incidentes separadas para homens e mulheres, bem como grupos de idade específicos, também foram calculados. Os números da população foram derivados dos dados do NSO. A taxa de prevalência foi estimada em 31 r de Dezembro de 2018. limites de confiança para incidência foram calculadas assumindo uma distribuição de Poisson.
Sequenciamento do genoma completo
A extração de DNA ocorreu de amostras de sangue venoso contendo EDTA inteiro usando o kit QIAamp DNA Mini QIAcube e a integridade do DNA foi medida usando o fluorômetro Quantus. O DNA foi sequenciado por todo o genoma pela plataforma BGISEQ-500 (BGI, Hong Kong, China) para gerar leituras de extremidades emparelhadas de 100 bp com uma profundidade média de 30 ×. As leituras foram alinhadas ao genoma de referência GRCh37 (HG19) usando o software Burrows – Wheeler Aligner. Variante de nucleotídeo único (SNV) e chamada de pequena inserção e exclusão (indel) e filtragem de qualidade foram realizadas usando o Genome Analysis Toolkit (GATK). A ferramenta ExpansionHunter foi usada para analisar tamanhos de repetição de ATXN2 (NM_002973.3: c.496_498CAG), C9orf72 (NM_001256054.2: c.-45 + 163GGGGCC) e NIPA1 (NM_144599.4: c.24_26GGC) [ 24] A fim de estimar a ancestralidade genética em relação aos mapas de referência de diversas populações, a análise de componentes principais (PCA) foi realizada em LASER com os resultados plotados usando a facilidade LASER Server plot ( https://laser.sph.umich.edu/ ) . HomozygosityMapper ( www.homozygositymapper.org ) [ 25 ] foi usado para mapear execuções de homozigosidade (ROHs).
Análise de variantes
Procuramos e analisamos as variantes e indels que codificam a proteína e que alteram o local de splice em 58 genes causadores ou de risco de ALS estabelecidos (Tabela 1 ). Restringimos as análises a variantes com frequência do alelo menor europeu (MAF) ≤0,01, que corresponde aproximadamente à frequência europeia do risco ALS recentemente descoberto NM_004984.2: c.2957C> T; p. (Pro986Leu) alelo no gene KIF5A [ 26 ]. Quando disponíveis, as variantes foram anotadas com informações do banco de dados dbSNP, incluindo estimativas de MAF específicas para a Europa do banco de dados de agregação do genoma (gnomAD). As frequências de alelos para casos e controles de ALS no conjunto de dados do Project MinE foram extraídas do banco de dados do Project MinE [ 27] Para determinar a patogenicidade das variantes, MetaSVM e MetaLR, dois métodos de predição baseados em conjunto integrando múltiplos sistemas de pontuação, foram usados em vista de sua capacidade preditiva superior em relação a outros métodos [ 28 ]. As variantes foram consideradas prejudiciais se os resultados de ambos os métodos coincidissem. Indels e variantes de aceitador / doador de site de splice foram automaticamente classificadas como deletérias. As variantes e os fenótipos associados foram submetidos ao banco de dados ClinVar ( https://www.ncbi.nlm.nih.gov/clinvar/ ) com os números de acesso SCV001426191, SCV001426206-SCV001426210, SCV001426221-SCV001426223 e SCV001437161-SCV001437192. Novas variantes detectadas na coorte caso-controle maltesa foram enviadas ao banco de dados dbSNP ( https://www.ncbi.nlm.nih.gov/snp/) com os IDs SNP de envio de ss2137544106, ss3986090479, ss3986090480 e ss3986090481.
Estatisticas
As comparações entre as médias foram feitas com o teste t de Student bicaudal não pareado, enquanto a comparação entre as variáveis categóricas foi feita com o teste χ 2 . Um valor de p <0,05 foi considerado significativo. Os dados foram processados com o software GraphPad Prism v8.4.0.
Resultados
Características base
As principais características dos pacientes e controles são detalhadas na Tabela 2 . Os casos de ALS eram predominantemente do sexo masculino, com início predominante dos sintomas na coluna vertebral. ALS de início bulbar teve uma ocorrência ligeiramente maior em mulheres (57,2%). O início da doença ocorreu por volta da meia-idade, com a mediana da idade de início sendo menor nas mulheres (59,5 anos) em comparação com os homens (64 anos). Apenas um paciente do sexo masculino e feminino tinha menos de 45 anos (8,3%). ALS precoce (≤55 anos) foi mais frequentemente espinhal no início (razão espinhal / bulbar = 9: 1). Uma história familiar de ELA foi registrada para uma minoria de casos (Tabela 2 ). No entanto, considerando sua agregação familiar com ALS [ 29 , 30 , 31], a inclusão de história familiar de demência e esquizofrenia ou psicose semelhante a endofenótipos neuropsiquiátricos aumenta a proporção de fALS para 37,5% (9/24 pacientes). A duração média da doença foi de 44,5 ± 28,3 DP meses, com os homens tendo uma progressão mais rápida em comparação com as mulheres (29,6 ± 8,8 DP meses vs. 66,8 ± 33,4 DP meses, p = 0,0065). O local de início da doença não influenciou a duração da doença (início espinhal = 43,5 ± 32,4 DP meses, início bulbar = 43,7 ± 19,8 DP meses, p = NS).
Um terço dos pacientes com ELA recrutados tinham uma história de tabagismo pesado e mais da metade relatou uma ocupação associada a atividades extenuantes, ambos os quais foram implicados como fatores de risco ambientais para ELA [ 32 , 33 ]. O abuso de álcool na coorte de pacientes foi mínimo. Visto que vários anos se passaram desde o início até o recrutamento, os níveis de CK na coorte de pacientes estavam, em média, apenas ligeiramente acima da faixa normal. A pontuação ALSFRS-R média em recrutas ALS foi quase metade da registrada para os indivíduos de controle com a pontuação no último próximo de 48, o máximo esperado em indivíduos saudáveis. A distribuição de casos e controles em todas as ilhas maltesas é exibida na Fig. 1. Uma maior densidade populacional no sudeste de Malta continental, muito provavelmente explica o aumento no número de casos nesta região geográfica em relação a outras regiões.
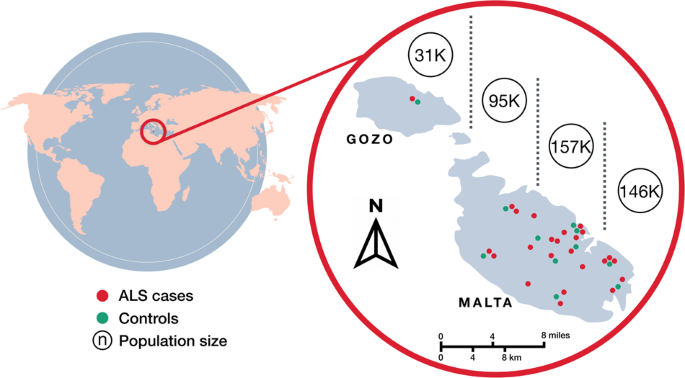
O tamanho da população das quatro divisões regionais (separadas por linhas pontilhadas) é baseado nos dados do NSO em 2017.
Taxas de incidência e prevalência
A taxa de incidência anual de ALS no período de 2017–2018 foi de 2,48 / 100.000 pessoas-ano (IC de 95% 1,59–3,68). A razão de incidência entre homens e mulheres foi de 1,93: 1. Portanto, a taxa de incidência foi maior para os homens (3,25, IC 95% 1,86–5,28) do que para as mulheres (1,68, IC 95% 0,72–3,31), e essa tendência ocorreu em todas as faixas etárias após 49 anos (Fig. 2 ). Tanto para homens quanto para mulheres, a incidência aumentou com a idade, mas diminuiu após os 79 anos. Os picos ocorreram na faixa de 50 a 59 anos entre os homens e na faixa de 70 a 79 anos entre as mulheres. Um total de 17 pacientes do sexo masculino (= 12, fêmea = 5) estavam vivos no dia prevalência (31 rDezembro de 2018), correspondendo a uma prevalência bruta de 3,44 / 100.000 (IC95% 2,01–5,52). Da mesma forma, a prevalência em homens (5,16, IC 95% 2,74–8,83) foi maior do que em mulheres (1,66, IC 95% 0,45–4,24), levando a uma razão de prevalência entre homens e mulheres de 3,11: 1. Não houve diferença na idade média de início e local de início entre os pacientes prevalentes e incidentes.
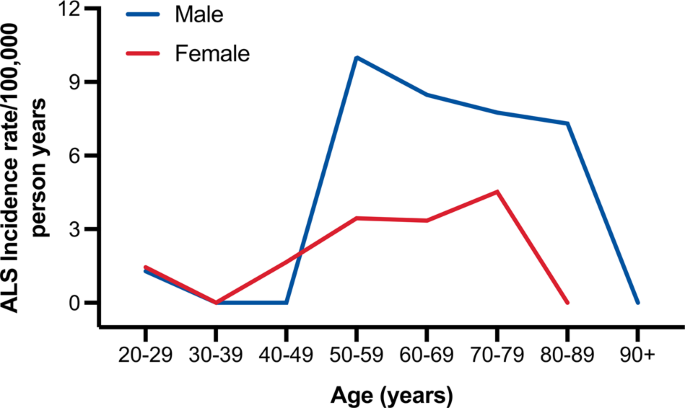
A taxa de incidência aumenta com a idade e foi maior para homens em comparação com mulheres em todas as faixas etárias após 49 anos. Os picos ocorrem na faixa etária de 50 a 59 anos para os homens e na faixa de 70 a 79 anos para as mulheres.
Ancestralidade genética
A análise de PCA da coorte de caso-controle maltesa mostrou que os casos e controles estavam dentro de 3 desvios padrão da média para as amostras combinadas ao longo dos componentes principais (PC) 1–4, garantindo assim a correspondência genética adequada. O mapeamento do paciente com ALS maltês e das amostras de controle para as coordenadas do PCA de referência para amostras do Painel de Diversidade do Genoma Humano (HGDP) mostra a sobreposição com a porção do cluster do Oriente Médio que faz fronteira com o cluster europeu (Fig. 3 ). Uma afinidade genética com as populações do Oriente Médio também é aparente nos sicilianos [ 34 , 35 ], apoiando ainda mais uma ancestralidade genética comum para as populações que habitam as ilhas mediterrâneas de Malta e Sicília.
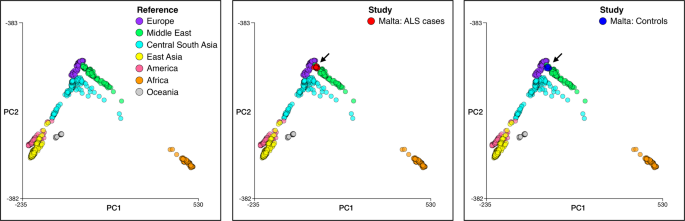
Painel esquerdo , coordenadas PCA de referência para amostras do painel de referência HGDP. Painel do meio , casos de ALS maltês mapeados para as coordenadas PCA de referência. Painel direito , controles malteses mapeados para as coordenadas PCA de referência.
Expansões de repetição em C9orf72 , ATXN2 e NIPA1 genes
Apesar dos estudos que mostram que C9orf72 é o principal gene que está mutado na população de ALS europeia [ 8 , 36 , 37 ], não identificamos expansões de repetição de hexanucleotídeo patogênica (GGGGCC) (≥24) em C9orf72 em casos de fALS ou sALS ( Fig. 4 ). O tamanho da repetição de expansão variou de 2 a 10 em controles e de 2 a 13 em pacientes com ELA. Consistente com estudos anteriores [ 36 , 37 ], um comprimento de repetição de 2 foi o mais predominante em ambos os grupos (42,3% nos controles e 52,2% nos casos de ELA). Além de C9orf72 , repita as expansões em outros genes, incluindo ATXN2 e NIPA1foram associados a risco aumentado de ALS [ 11 , 38 ]. Identificamos um paciente do sexo masculino com fALS que possuía expansões de repetição de trinucleotídeos associadas a ATXN2 ALS (28 repetições de comprimento) no estado homozigoto (Fig. 4 ). Aos 67 anos, esse paciente apresentou pela primeira vez fraqueza bilateral nas pernas, que progrediu. Ele subsequentemente desenvolveu disartria e sucumbiu à doença 2 anos após o início da doença. Esse paciente também foi o único em nossa coorte que apresentou sinais de comprometimento cognitivo. A história familiar era notável para uma irmã falecida que tinha ALS com início precoce no final dos 30 anos e uma mãe falecida que tinha demência (amostras de DNA não estavam disponíveis para estudo). O pedigree é mostrado na Fig. Complementar. S1 . O tamanho máximo de repetição ATXN2 observado em controles saudáveis foi de 25, encontrado no estado heterozigoto, em um sujeito. Todos os pacientes e controles restantes com ELA tiveram comprimentos de repetição ≤23. Como foi relatado anteriormente [ 11 , 39 ], um comprimento de repetição de 22, detectado principalmente no estado homozigoto, foi o alelo ATXN2 mais abundante (88,5% em controles e 84,8% em pacientes com ELA). Considerando NIPA1 , encontramos um paciente do sexo masculino com sALS que teve um [ 21] Motivo de repetição GCG no estado heterozigoto. O paciente teve início tardio (73 anos), apresentando fraqueza bilateral primeiro nos membros inferiores e depois nos superiores. A sobrevida foi mais curta (<2,5 anos) em comparação com a sobrevida mediana (3,5 anos) em nossa coorte de ALS. Nos controles e nos casos restantes de ALS, o comprimento da repetição foi variável variando de 5 a 13. Semelhante aos achados anteriores [ 38 , 40 ], os alelos mais frequentes consistiam em 7 ou 8 repetições, com as respectivas frequências alélicas sendo 26,1% e 67,4 % em pacientes com ELA e 7,7% e 73,1% nos controles.
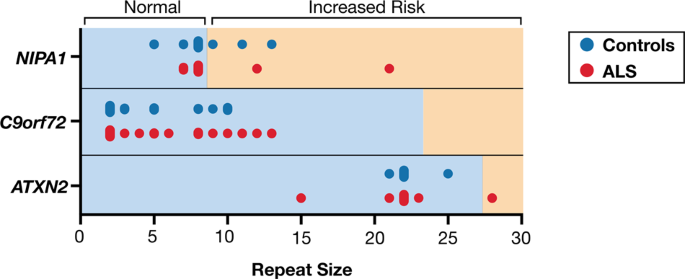
Gráfico de dispersão mostrando distribuição e frequência de tamanhos de repetição, indicados por círculos.
Variantes genéticas em genes associados a ALS conhecidos
Curiosamente, a coorte de pacientes com ALS maltês foi considerada negativa para SNVs não sinônimos ou que alteram o local de splice nos genes SOD1 , TARDBP ou FUS , que são os genes ALS mais comumente mutados, nessa ordem, seguindo C9orf72 [ 8 ]. Depois de examinar 58 genes associados à ELA em nosso paciente e coorte de controle, identificamos 35 variantes de codificação raras (MAF europeia ≤ 0,01) que estavam presentes em pacientes malteses com ELA e ausentes em controles (Tabela 3 ). Três SNVs em DDX20 , EWSR1 ou GLE1 não foram encontrados no banco de dados dbSNP (v141). O NM_001003722.1: c.2078C> T; p. (Ser693Phe) variante em GLE1foi no entanto relatado em um estudo recente como uma mutação fundadora em maltês que na homozigose induz uma síndrome de disfunção motora que se apresenta na infância [ 21 ]. Variantes previstas para serem prejudiciais por MetaSVM e MetaLR foram detectadas em ALS2 , DAO , DCTN1 , ERBB4 , SCFD1 e SPG11 (Tabela 3 ). Todos foram detectados em pacientes com sALS. O NM_001917.4: c.250G> A; A variante p. (Ala84Thr) em DAO foi detectada em dois pacientes, enquanto uma paciente do sexo feminino possuía variantes deletérias em mais de um gene ( DAO e DCTN1 ).
A análise dos indels permitiu identificar uma deleção rara em SETX , detectada no estado heterozigoto em um caso de sALS (Tabela 3 ). Esse paciente apresentou fraqueza nos membros superiores aos 70 anos. Um ano depois, no acompanhamento, a fraqueza se espalhou para os membros inferiores. Prevê-se que a deleção resulte em uma mudança de quadro, conseqüentemente produzindo uma proteína SETX truncada sem o domínio helicase. É digno de nota que as variantes prejudiciais ALS2 , SCFD1 e SETX detectadas em pacientes malteses com sALS apresentaram frequências alélicas mais altas em pacientes com ALS no conjunto de dados caso-controle do Project MinE [ 27 ], ressaltando assim sua provável patogenicidade (Tabela 3 ). SNVs ou indels raros que eram exclusivos dos controles ou que eram compartilhados por pacientes e controles com ELA estão listados na Tabela Suplementar S1 . Um SNVs na FIG4 não foi encontrado no banco de dados dbSNP (v141). Nenhum alongamento homozigoto foi superrepresentado em pacientes com sALS em comparação com os controles (Fig. Suplementar S2 ). Para estimar o risco genético de ALS na população maltesa, determinamos que a porcentagem de casos de sALS causados por variantes raras e potencialmente deletérias (ausentes nos controles) em pelo menos um gene associado à ALS foi de 40% (8/20 pacientes). Dois casos de FALS não carregavam nenhuma mutação em genes de ALS conhecidos ou loci de risco, portanto, justificando mais estudos para elucidar novos genes que causam ALS.
Discussão
Em nosso trabalho, investigamos as características dos pacientes malteses com ELA, descrevemos seu perfil genético e determinamos a incidência e prevalência de ELA nas ilhas maltesas. É interessante que os aspectos específicos da população de nossos casos de ALS se sobreponham aos relatados para outras populações europeias vizinhas, especialmente aquelas no Mediterrâneo, incluindo a ilha da Sicília [ 41 ] e a região sul de Puglia [ 42 , 43 ] na Itália, Tunísia [ 44 ] e Chipre [ 45 ]. Embora a preponderância masculina em ALS seja virtualmente universal [ 46 , 47], é digno de nota que nessas populações específicas, bem como em Malta, a idade de início da doença é maior em homens do que em mulheres. Isso está em contraste com as populações do norte da bacia do Mediterrâneo, incluindo Catalunha na Espanha [ 48 ], e as regiões de Emilia Romagna [ 49 ], Ligúria [ 50 ] ou Friuli-Venezia-Giulia [ 51 ] na Itália. A incidência e prevalência de ALS em Malta é semelhante à mediana europeia [ 3 ].
Relatamos uma porcentagem maior (12,5%) de casos de FALS em Malta, próximo ao relatado para a população da Ligúria no norte da Itália (10%) [ 50 ], mas quase metade do relatado para a ilha da Sardenha (26,7%) [ 52 ] . No entanto, semelhante a estudos anteriores [ 53 ], relaxar os critérios rigorosos, incluindo condições neurológicas em famílias que têm uma sobreposição genética com ALS [ 54 ], pode aumentar a porcentagem de FALS em Malta em três vezes (37,5%). Em concordância, considerando o subconjunto de sALS maltês, mostramos que, em comparação com outras populações europeias [ 10 ] ou populações de ascendência europeia [ 9 ], uma proporção maior de pacientes aparentemente sALS é provavelmente determinada geneticamente.
Curiosamente, variantes raras deletérias nos principais genes de ALS, incluindo C9orf72 , SOD1 , TARDBP e FUS , estavam ausentes em pacientes malteses com ALS. Esta descoberta confirma a presença de um gradiente Norte-Sul na frequência de mutações dentro desses genes em toda a Europa. Conseqüentemente, as mutações C9orf72 ou SOD1 em fALS são mais altas em países do norte da Europa, como Bélgica e Finlândia, enquanto uma frequência relativamente baixa é registrada no sul da Europa, incluindo Espanha e Itália continental [ 8 ]. Uma situação semelhante pode ser observada para TARDBP e FUS [ 8] Nosso estudo, portanto, ressalta as diferenças marcantes que existem entre grupos étnicos e regiões geográficas no que diz respeito aos genes que estão comumente implicados na ELA.
Pacientes malteses com ALS, no entanto, possuíam alelos deletérios em genes 'menores' de ALS, incluindo ALS2 , ATXN2 , DAO , DCTN1 , ERBB4 , NIPA1 , SETX , SCFD1 e SPG11 . ALS2 e SPG11 foram associados com ALS de início juvenil apenas sob um modelo de doença recessiva [ 55 , 56 , 57 , 58 ]. Neste contexto, uma vez que observamos variantes no ALS2 e SPG11genes apenas em configurações heterozigotas e em pacientes com ELA de início na idade adulta, é provável que esses alelos não fossem causadores de doença nos pacientes que os possuíam. No entanto, considerando que o ALS tem uma base oligogênica [ 59 ], um efeito modificador ou aditivo não pode ser excluído. O mesmo pode ser dito para SCFD1 , que só foi recentemente identificado como um locus de risco [ 13 ], e para o qual relatamos uma variante prejudicial em um paciente com ELA com início de idade jovem (27 anos) e cuja progressão da doença é excepcionalmente lento.
ATXN2 , DAO , DCTN1 , ERBB4 , NIPA1 e SETX foram previamente associados com ALS tendo um modo autossômico dominante de herança [ 11 , 38 , 40 , 60 , 61 , 62 , 63 ]. Alelos prejudiciais descobertos em nossa coorte de pacientes, que visam especificamente esses genes, são provavelmente os causadores. É interessante notar que o ERBB4c.3814G> A; p. (Gly1272Arg) variante relatada neste estudo é extremamente próxima daquela relatada em um indivíduo japonês com sALS [c.3823C> T; p. (Arg1275Trp)], ambos os quais estão localizados no domínio C-terminal da proteína, perto de vários locais de fosforilação, que medeiam as vias de sinalização a jusante [ 61 ]. Embora as variantes SETX tenham sido descobertas inicialmente em pacientes com ELA de início juvenil [ 62 ], relatórios têm descrito alelos prejudiciais em pacientes com ELA de início na idade adulta [ 10 , 64 ]. Isso está de acordo com nosso estudo, portanto, o paciente maltês com ELA possuindo uma deleção SETX teve uma idade de início tardia semelhante à relatada em um estudo de caso anterior [ 64]
Nossas descobertas têm implicações importantes. A incidência e a prevalência de ALS em Malta, bem como os aspectos da população de pacientes, se sobrepõem aos de países vizinhos. No entanto, apoiado por resultados de ancestralidade genética, a arquitetura genética de ALS em Malta parece ser diferente da média europeia, sublinhando o isolamento genético imposto pela geografia. Isso, combinado com a falta de um fator genético identificado em dois terços dos casos de fALS em Malta, incentiva mais estudos com o objetivo de descobrir novos genes de ALS. Nossos dados "preliminares" excluem a possibilidade de que esses pacientes tenham variantes deletérias em um conjunto de genes associados a outros distúrbios do neurônio motor, incluindo ataxias hereditárias e neuropatias motoras e sensoriais hereditárias (dados não mostrados). Finalmente,as variantes descritas neste trabalho devem estimular a geração de modelos animais para confirmar a causa e entender melhor os mecanismos da doença [65 ]. Isso é imperativo especialmente para genes ALS 'menores', uma vez que eles são relativamente menos estudados do que genes 'principais', mas que são conseqüentes em populações específicas.
Referências
- 1
Brown RH, Al-Chalabi A. Esclerose lateral amiotrófica. N Engl J Med. 2017; 377: 162–72.
- 2
van Es MA, Hardiman O, Chio A, Al-Chalabi A, Pasterkamp RJ, Veldink JH, et al. Esclerose lateral amiotrófica. Lanceta. 2017; 390: 2084–98.
- 3
Chio A, Logroscino G, Traynor BJ, Collins J, Simeone JC, Goldstein LA, et al. Epidemiologia global da esclerose lateral amiotrófica: uma revisão sistemática da literatura publicada. Neuroepidemiologia. 2013; 41: 118–30.
- 4
Byrne S, Walsh C, Lynch C, Bede P, Elamin M, Kenna K, et al. Taxa de esclerose lateral amiotrófica familiar: uma revisão sistemática e meta-análise. J Neurol Neurosurg Psychiatry. 2011; 82: 623–7.
- 5
Al-Chalabi A, Fang F, Hanby MF, Leigh PN, Shaw CE, Ye W, et al. Uma estimativa da herdabilidade da esclerose lateral amiotrófica usando dados de gêmeos. J Neurol Neurosurg Psychiatry. 2010; 81: 1324–6.
- 6
Ryan M, Heverin M, McLaughlin RL, Hardiman O. Risco ao longo da vida e herdabilidade da esclerose lateral amiotrófica. JAMA Neurol. 2019; 76: 1367–74.
- 7
Trabjerg BB, Garton FC, van Rheenen W., Fang F, Henderson RD, Mortensen PB, et al. ALS em registros dinamarqueses: herdabilidade e links para transtornos psiquiátricos e cardiovasculares. Neurol Genet. 2020; 6: e398.
- 8
Zou ZY, Zhou ZR, Che CH, Liu CY, He RL, Huang HP. Epidemiologia genética da esclerose lateral amiotrófica: uma revisão sistemática e meta-análise. J Neurol Neurosurg Psychiatry. 2017; 88: 540–9.
- 9
Gibson SB, Downie JM, Tsetsou S, Feusier JE, Figueroa KP, Bromberg MB, et al. O risco genético em evolução para ELA esporádica. Neurologia. 2017; 89: 226–33.
- 10
Kenna KP, McLaughlin RL, Byrne S, Elamin M, Heverin M, Kenny EM, et al. Delineando a heterogeneidade genética de ALS usando sequenciamento de alto rendimento direcionado. J Med Genet. 2013; 50: 776–83.
- 11
Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, et al. As expansões de poliglutamina de comprimento intermediário da Ataxina-2 estão associadas a um risco aumentado de ELA. Natureza. 2010; 466: 1069–75.
- 12
van Es MA, Veldink JH, Saris CG, Blauw HM, van Vught PW, Birve A, et al. O estudo de associação de todo o genoma identifica 19p13.3 (UNC13A) e 9p21.2 como loci de suscetibilidade para esclerose lateral amiotrófica esporádica. Nat Genet. 2009; 41: 1083–7.
- 13
van Rheenen W., Shatunov A., Dekker AM, McLaughlin RL, Diekstra FP, Pulit SL, et al. As análises de associação de todo o genoma identificam novas variantes de risco e a arquitetura genética da esclerose lateral amiotrófica. Nat Genet. 2016; 48: 1043–8.
- 14
Taylor JP, Brown RH Jr., Cleveland DW. Decodificando ALS: dos genes ao mecanismo. Natureza. 2016; 539: 197–206.
- 15
Ly CV, Miller TM. Novos oligonucleotídeos antisense e terapias virais para esclerose lateral amiotrófica. Curr Opin Neurol. 2018; 31: 648–54.
- 16
Arcos-Burgos M, Muenke M. Genética de isolados de população. Clin Genet. 2002; 61: 233–47.
- 17
Caruana J. Population genetics of Western Mediterranean Islands — Malta: a case study. Manchester: Universidade de Manchester; 2012
- 18
Capelli C, Redhead N, Romano V, Cali F, Lefranc G, Delague V, et al. Estrutura populacional na bacia do Mediterrâneo: uma perspectiva do cromossomo Y. Ann Hum Genet. 2006; 70: 207–25.
- 19
Cassar M, Farrugia C, Vidal C. Frequências alélicas de 14 loci STR na população de Malta. Leg Med. 2008; 10: 153–6.
- 20
Borg J, Papadopoulos P, Georgitsi M, Gutierrez L, Grech G, Fanis P, et al. A haploinsuficiência para o fator de transcrição eritróide KLF1 causa persistência hereditária da hemoglobina fetal. Nat Genet. 2010; 42: 801–5.
- 21
Dito E, Chong JX, Hempel M, Denecke J, Soler P, Strom T, et al. A sobrevivência além do período perinatal expande os fenótipos causados por mutações em GLE1. Am J Med Genet A. 2017; 173: 3098–103.
- 22
Brooks BR, Miller RG, Swash M, Munsat TL. Grupo de Pesquisa da Federação Mundial de Neurologia em Neurônio Motor D. El Escorial revisitado: critérios revisados para o diagnóstico de esclerose lateral amiotrófica. Amyotroph Lateral Scler Outro Mot Neuron Disord. 2000; 1: 293–9.
- 23
Ludolph A, Drory V, Hardiman O, Nakano I, Ravits J, Robberecht W, et al. Uma revisão dos critérios do El Escorial - 2015. Amyotroph Lateral Scler Frontotemporal Degener. 2015; 16: 291–2.
- 24
Dolzhenko E, van Vugt J, Shaw RJ, Bekritsky MA, van Blitterswijk M, Narzisi G, et al. Detecção de expansões repetidas longas a partir de dados de sequência do genoma completo sem PCR. Genome Res. 2017; 27: 1895–903.
- 25
Seelow D, Schuelke M. HomozygosityMapper2012-colmatando a lacuna entre o mapeamento de homozigosidade e o sequenciamento profundo. Nucleic Acids Res. 2012; 40: W516–20.
- 26
Nicolas A, Kenna KP, Renton AE, Ticozzi N, Faghri F, Chia R, et al. As análises de todo o genoma identificam KIF5A como um novo gene ALS. Neuron. 2018; 97: 1268–83 e6.
- 27
van der Spek RAA, van Rheenen W., Pulit SL, Kenna KP, van den Berg LH, Veldink JH, et al. O projeto MinE databrowser: trazendo o sequenciamento de todo o genoma em larga escala em ALS para pesquisadores e o público. Amyotroph Lateral Scler Frontotemporal Degener. 2019; 20: 432–40.
- 28
Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, et al. Comparação e integração de métodos de predição de deletérios para SNVs não sinônimos em estudos de sequenciamento de exoma completo. Hum Mol Genet. 2015; 24: 2125–37.
- 29
Byrne S, Heverin M, Elamin M, Bede P, Lynch C, Kenna K, et al. Agregação de doenças neurológicas e neuropsiquiátricas em famílias com esclerose lateral amiotrófica: um estudo de coorte caso-controle de base populacional de esclerose lateral amiotrófica familiar e esporádica. Ann Neurol. 2013; 74: 699–708.
- 30
O'Brien M., Burke T., Heverin M., Vajda A., McLaughlin R., Gibbons J, et al. Agrupamento de doenças neuropsiquiátricas em parentes de primeiro e segundo grau de pacientes com esclerose lateral amiotrófica. JAMA Neurol. 2017; 74: 1425–30.
- 31
Longinetti E, Mariosa D, Larsson H, Ye W, Ingre C, Almqvist C, et al. Doenças neurodegenerativas e psiquiátricas em famílias com esclerose lateral amiotrófica. Neurology 2017; 89: 578–85.
- 32
Peters S, Visser AE, D'Ovidio F, Vlaanderen J, Portengen L, Beghi E, et al. Modificação do efeito da associação entre o tabagismo total e risco de ALS por intensidade, duração e tempo desde o abandono: Euro-MOTOR. J Neurol Neurosurg Psychiatry. 2020; 91: 33–9.
- 33
Visser AE, Rooney JPK, D'Ovidio F, Westeneng HJ, Vermeulen RCH, Beghi E, et al. Estudo multicêntrico, transcultural, de base populacional e caso-controle da atividade física como fator de risco para esclerose lateral amiotrófica. J Neurol Neurosurg Psychiatry. 2018; 89: 797–803.
- 34
Di Gaetano C, Voglino F, Guarrera S, Fiorito G, Rosa F, Di Blasio AM, et al. Uma visão geral da estrutura genética dentro da população italiana a partir de dados de todo o genoma. PLoS ONE. 2012; 7: e43759.
- 35
Sarno S, Boattini A, Pagani L, Sazzini M, De Fanti S, Quagliariello A, et al. Camadas de mistura antigas e recentes na Sicília e no sul da Itália traçam múltiplas rotas de migração ao longo do Mediterrâneo. Sci Rep. 2017; 7: 1984.
- 36
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. Uma expansão de repetição de hexanucleotídeo em C9ORF72 é a causa do ALS-FTD ligado ao cromossomo 9p21. Neuron. 2011; 72: 257–68.
- 37
Smith BN, Newhouse S, Shatunov A, Vance C, Topp S, Johnson L, et al. A mutação de expansão C9ORF72 é uma causa comum de ALS +/- FTD na Europa e tem um único fundador. Eur J Hum Genet. 2013; 21: 102–8.
- 38
Blauw HM, van Rheenen W., Koppers M., Van Damme P., Waibel S, Lemmens R., et al. As expansões de repetição de polialanina NIPA1 estão associadas à esclerose lateral amiotrófica. Hum Mol Genet. 2012; 21: 2497–502.
- 39
Van Damme P, Veldink JH, van Blitterswijk M, Corveleyn A, van Vught PW, Thijs V, et al. O tamanho de repetição ATXN2 CAG expandido em ALS identifica a sobreposição genética entre ALS e SCA2. Neurologia. 2011; 76: 2066–72.
- 40
Tazelaar GHP, Dekker AM, van Vugt J, van der Spek RA, Westeneng HJ, Kool L, et al. Associação de expansões de repetição NIPA1 com esclerose lateral amiotrófica em uma grande coorte internacional. Neurobiol Aging. 2019; 74: 234 e9–15.
- 41
Ragonese P, Cellura E, Aridon P, D'Amelio M, Spataro R, Taiello AC, et al. Incidência de esclerose lateral amiotrófica na Sicília: um estudo de base populacional. Amyotroph Lateral Scler. 2012; 13: 284–7.
- 42
Logroscino G, Beghi E, Zoccolella S, Palagano R, Fraddosio A, Simone IL, et al. Incidência de esclerose lateral amiotrófica no sul da Itália: um estudo de base populacional. J Neurol Neurosurg Psychiatry. 2005; 76: 1094–8.
- 43
Zoccolella S, Beghi E, Palagano G, Fraddosio A, Samarelli V, Lamberti P, et al. Sinais e sintomas no diagnóstico de esclerose lateral amiotrófica: um estudo de base populacional no sul da Itália. Eur J Neurol. 2006; 13: 789–92.
- 44
Kacem I, Sghaier I, Bougatef S, Nasri A, Gargouri A, Ajroud-Driss S, et al. Aspectos epidemiológicos e clínicos da esclerose lateral amiotrófica em uma coorte tunisiana. Amyotroph Lateral Scler Frontotemporal Degener. 2020; 21: 131–9.
- 45
Demetriou CA, Hadjivasiliou PM, Kleopa KA, Christou YP, Leonidou E, Kyriakides T, et al. Epidemiologia da esclerose lateral amiotrófica na República de Chipre: um estudo retrospectivo de 25 anos. Neuroepidemiologia. 2017; 48: 79–85.
- 46
Logroscino G, Traynor BJ, Hardiman O, Chio A, Mitchell D, Swingler RJ, et al. Incidência de esclerose lateral amiotrófica na Europa. J Neurol Neurosurg Psychiatry. 2010; 81: 385–90.
- 47
Marin B, Boumediene F, Logroscino G, Couratier P, Babron MC, Leutenegger AL, et al. Variação na incidência mundial de esclerose lateral amiotrófica: uma meta-análise. Int J Epidemiol. 2017; 46: 57–74.
- 48
Pradas J, Puig T, Rojas-Garcia R, Viguera ML, Gich I, Logroscino G, et al. Esclerose lateral amiotrófica na Catalunha: um estudo de base populacional. Amyotroph Lateral Scler Frontotemporal Degener. 2013; 14: 278–83.
- 49.
Mandrioli J, Biguzzi S, Guidi C, Venturini E, Sette E, Terlizzi E, et al. Epidemiologia da esclerose lateral amiotrófica na região de Emilia Romagna (Itália): um estudo de base populacional. Amyotroph Lateral Scler Frontotemporal Degener. 2014; 15: 262–8.
- 50
Scialo C, Novi G, Bandettini di Poggio M, Canosa A, Sormani MP, Mandich P. et al. Epidemiologia clínica da esclerose lateral amiotrófica na Ligúria, Itália: uma atualização do registro LIGALS. Amyotroph Lateral Scler Frontotemporal Degener. 2016; 17: 535–42.
- 51
Palese F, Sartori A, Verriello L, Ros S, Passadore P, Manganotti P, et al. Epidemiologia da esclerose lateral amiotrófica em Friuli-Venezia Giulia, Nordeste da Itália, 2002-2014: um estudo retrospectivo de base populacional. Amyotroph Lateral Scler Frontotemporal Degener. 2019; 20: 90–9.
- 52
Borghero G, Pugliatti M, Marrosu F, Marrosu MG, Murru MR, Floris G, et al. Arquitetura genética de ALS na Sardenha. Neurobiol Aging. 2014; 35: 2882 e7–12.
- 53
Ryan M., Heverin M., Doherty MA, Davis N., Corr EM, Vajda A, et al. Determinando a incidência de familialidade em ALS: Um estudo de tendências temporais na Irlanda de 1994 a 2016. Neurol Genet. 2018; 4: e239.
- 54
McLaughlin RL, Schijven D, van Rheenen W., van Eijk KR, O'Brien M, Kahn RS, et al. Correlação genética entre esclerose lateral amiotrófica e esquizofrenia. Nat Commun. 2017; 8: 14774.
- 55
Hadano S, Hand CK, Osuga H, Yanagisawa Y, Otomo A, Devon RS, et al. Um gene que codifica um regulador GTPase putativo sofre mutação na esclerose lateral amiotrófica familiar 2. Nat Genet. 2001; 29: 166–73.
- 56
Hentati A, Bejaoui K, Pericak-Vance MA, Hentati F, Speer MC, Hung WY, et al. Ligação da esclerose lateral amiotrófica familiar recessiva ao cromossomo 2q33-q35. Nat Genet. 1994; 7: 425–8.
- 57
Daoud H, Zhou S, Noreau A, Sabbagh M, Belzil V, Dionne-Laporte A, et al. O sequenciamento do exoma revela mutações SPG11 causando ALS juvenil. Neurobiol Aging. 2012; 33: 839 e5–9.
- 58
Orlacchio A, Babalini C, Borreca A, Patrono C, Massa R, Basaran S, et al. Mutações no SPATACSIN causam esclerose lateral amiotrófica juvenil autossômica recessiva. Cérebro. 2010; 133: 591–8.
- 59.
van Blitterswijk M, van Es MA, Hennekam EA, Dooijes D, van Rheenen W, Medic J, et al. Evidência de uma base oligogênica da esclerose lateral amiotrófica. Hum Mol Genet. 2012; 21: 3776–84.
- 60
Mitchell J, Paul P, Chen HJ, Morris A, Payling M, Falchi M, et al. A esclerose lateral amiotrófica familiar está associada a uma mutação na D-aminoácido oxidase. Proc Natl Acad Sei USA. 2010; 107: 7556–61.
- 61
Takahashi Y, Fukuda Y, Yoshimura J, Toyoda A, Kurppa K, Moritoyo H, et al. Mutações ERBB4 que interrompem a via neuregulina-ErbB4 causam esclerose lateral amiotrófica tipo 19. Am J Hum Genet. 2013; 93: 900–5.
- 62
Chen YZ, Bennett CL, Huynh HM, Blair IP, Puls I, Irobi J, et al. Mutações do gene da helicase DNA / RNA em uma forma de esclerose lateral amiotrófica juvenil (ALS4). Am J Hum Genet. 2004; 74: 1128–35.
- 63
Munch C, Sedlmeier R, Meyer T, Homberg V, Sperfeld AD, Kurt A, et al. Mutações pontuais da subunidade p150 do gene da dinactina (DCTN1) em ALS. Neurologia. 2004; 63: 724–6.
- 64
Tripolszki K, Torok D, Goudenege D, Farkas K, Sulak A, Torok N, et al. O sequenciamento de alto rendimento revelou uma nova mutação SETX em um paciente húngaro com esclerose lateral amiotrófica. Brain Behav. 2017; 7: e00669.
- 65
Aquilina B, Cauchi RJ. Modelagem de doenças do neurônio motor em moscas-das-frutas: lições da atrofia muscular espinhal. J Neurosci Methods. 2018; 310: 3–11.
Reconhecimentos
Os autores agradecem ao Prof. Richard Muscat, Wilfred Kenely e à Dra. Graziella Zahra pelo apoio a este trabalho. Agradecemos também a Matthew Camilleri pelo apoio técnico e administrativo inabalável. Continuamos gratos a todos os participantes deste estudo.
Financiamento
Este trabalho foi financiado pelo Fundo de Excelência em Pesquisa da Universidade de Malta para o RJC, e pelo Prêmio de Parceria de Internacionalização do Conselho de Malta para Ciência e Tecnologia para o RJC. A RB foi apoiada por uma bolsa de curta duração EMBO e por uma Bolsa Bjorn Formosa para Pesquisa Avançada em ALS / MND financiada pela organização sem fins lucrativos, ALS Malta Foundation, facilitada pelo Research Trust (RIDT) da Universidade de Malta. O ML foi apoiado por uma bolsa Endeavor (Malta), co-financiada pela UE - Fundo Social Europeu no âmbito do Programa Operacional II - Política de Coesão 2014-2020, “Investir no capital humano para criar mais oportunidades e promover o bem-estar da sociedade” .
Informação sobre o autor
Afiliações
autor correspondente
Declarações de ética
Conflito de interesses
Os autores declaram não haver conflito de interesses.
Informação adicional
Nota do editor A Springer Nature permanece neutra em relação a reivindicações jurisdicionais em mapas publicados e afiliações institucionais.
Informação suplementar
Direitos e permissões
Acesso aberto Este artigo está licenciado sob uma Licença Internacional Creative Commons Atribuição 4.0, que permite o uso, compartilhamento, adaptação, distribuição e reprodução em qualquer meio ou formato, desde que você dê o devido crédito ao (s) autor (es) original (is) e à fonte, fornecer um link para a licença Creative Commons e indicar se foram feitas alterações. As imagens ou outro material de terceiros neste artigo estão incluídos na licença Creative Commons do artigo, a menos que indicado de outra forma em uma linha de crédito para o material. Se o material não estiver incluído na licença Creative Commons do artigo e seu uso pretendido não for permitido por regulamentação legal ou exceder o uso permitido, você precisará obter permissão diretamente do detentor dos direitos autorais. Para ver uma cópia desta licença, visitehttp://creativecommons.org/licenses/by/4.0/ .
Nenhum comentário:
Postar um comentário
Observação: somente um membro deste blog pode postar um comentário.