Datação por carbono é recalibrada e deve mudar idade dos fósseis

Imagem: The Nanjing Institute/Divulgação
Desde
sua criação, em 1960, a datação por radiocarbono é usada para
determinar a idade de qualquer coisa, de livros e esqueletos a múmias e
marfim traficado. Agora, esse sistema será recalibrado com a inclusão de
milhares de dados obtidos desde sua última versão, de 2013.
“Uma nova curva de calibração é de importância fundamental para a compreensão da pré-história e da cronologia dos hominídeos", diz o cronologista arqueológico e diretor da Unidade de Acelerador de Radiocarbono de Oxford, Tom Higham.
Esse ajuste está sendo feito não apenas para melhor entender o passado, mas para compensar o presente, que hoje despeja bilhões de toneladas de carbono no meio ambiente. A queima de combustíveis fósseis e os testes de bombas nucleares mudaram radicalmente a quantidade de carbono-14 no ar.
A datação por radiocarbono pode dizer quando o homem moderno surgiu.Fonte: Science Photos/Reprodução
Quando uma planta ou um animal morre, para de absorver esse isótopo, e o que foi acumulado começa a decair. Medir a quantidade restante fornece uma estimativa de há quanto tempo algo morreu.
O problema é que a humanidade, principalmente nas últimas décadas, tem lançado na atmosfera o carbono de combustíveis fósseis, que não têm mais uma quantidade mensurável de carbono-14. O resultado é que bilhões de toneladas de carbono-12 artificialmente postas na balança estão arruinando a proporção constante entre os dois isótopos.

Os anéis das árvores ajudam a calibrar a curva de datação por radiocarbono.Fonte: Flickr/Tina Negus/Reprodução
Para produzir uma curva que possa ser usada para relacionar anos do calendário com os determinados pelo radiocarbono, é necessária uma sequência de amostras com data seguramente conhecida, como os anéis no tronco de uma árvore. Os cientistas usam esse método combinado com informações de sedimentos de lagos e oceanos, corais e estalagmites para calibrar o sistema.
Desde 1998 é usada o sistema IntCal (IntCal98), que já sofreu três atualizações: em 2004, 2009 e 2013. Agora, está sendo lançada a versão 2020, com as novas curvas para os hemisfério Norte (IntCal20) e sul (SHCal20) e mais para amostras marinhas (MarineCal20).
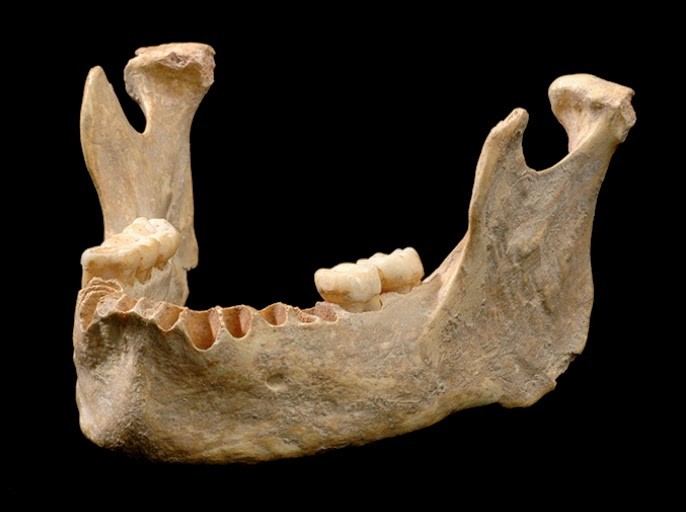
A mandíbula do Osase 1, o homem moderno mais antigo já encontrado.Fonte: FU Qiaomei/Phys.org/Reprodução
Ali foram escavados os restos mais antigos de humanos modernos (entre 35 e 40 milhares de anos). A idade do exemplar denominado Oase 1 foi reavaliada pela nova curva; ele é centenas de anos mais velho do que se pensava. Análises genéticas ainda revelaram que ele tinha um ancestral neandertal, distante de quatro a seis gerações.
“Isso significa que, quanto mais antigo o Oase 1 é, mais neandertais andavam pela Europa”, explica Higham. Em contrapartida, o adolescente cujos fósseis foram desenterrados na Sibéria viveu mil anos à frente do que se pensava e isso, segundo o cronólogo, muda a história da chegada dos humanos modernos na Sibéria.a
“Uma nova curva de calibração é de importância fundamental para a compreensão da pré-história e da cronologia dos hominídeos", diz o cronologista arqueológico e diretor da Unidade de Acelerador de Radiocarbono de Oxford, Tom Higham.
Esse ajuste está sendo feito não apenas para melhor entender o passado, mas para compensar o presente, que hoje despeja bilhões de toneladas de carbono no meio ambiente. A queima de combustíveis fósseis e os testes de bombas nucleares mudaram radicalmente a quantidade de carbono-14 no ar.
A datação por radiocarbono pode dizer quando o homem moderno surgiu.Fonte: Science Photos/Reprodução
Poluição altera o equilíbrio
Quase 99% de todo o carbono da Terra está na forma de seu isótopo com 12 nêutrons em seu núcleo; o carbono-12 é a forma mais estável do elemento. A técnica de datação por radiocarbono mede a presença de um isótopo instável e por isso, muito mais raro: o carbono-14 que, na natureza, é produzido a uma taxa constante na atmosfera superior da Terra.Quando uma planta ou um animal morre, para de absorver esse isótopo, e o que foi acumulado começa a decair. Medir a quantidade restante fornece uma estimativa de há quanto tempo algo morreu.
O problema é que a humanidade, principalmente nas últimas décadas, tem lançado na atmosfera o carbono de combustíveis fósseis, que não têm mais uma quantidade mensurável de carbono-14. O resultado é que bilhões de toneladas de carbono-12 artificialmente postas na balança estão arruinando a proporção constante entre os dois isótopos.
Árvores guardam um calendário confiável
Os anéis das árvores ajudam a calibrar a curva de datação por radiocarbono.Fonte: Flickr/Tina Negus/Reprodução
Para produzir uma curva que possa ser usada para relacionar anos do calendário com os determinados pelo radiocarbono, é necessária uma sequência de amostras com data seguramente conhecida, como os anéis no tronco de uma árvore. Os cientistas usam esse método combinado com informações de sedimentos de lagos e oceanos, corais e estalagmites para calibrar o sistema.
Desde 1998 é usada o sistema IntCal (IntCal98), que já sofreu três atualizações: em 2004, 2009 e 2013. Agora, está sendo lançada a versão 2020, com as novas curvas para os hemisfério Norte (IntCal20) e sul (SHCal20) e mais para amostras marinhas (MarineCal20).
Mais velho, mais jovem
A IntCal20 é baseada em 12.904 pontos de dados, quase o dobro do tamanho do conjunto de dados de 2013. Isso tornou, por exemplo, mais acurada a datação de um maxilar do Homo sapiens encontrado em 2002 na Pe?tera cu Oase (Ou a Caverna com Ossos), na Romênia. A mandíbula do Osase 1, o homem moderno mais antigo já encontrado.Fonte: FU Qiaomei/Phys.org/Reprodução
Ali foram escavados os restos mais antigos de humanos modernos (entre 35 e 40 milhares de anos). A idade do exemplar denominado Oase 1 foi reavaliada pela nova curva; ele é centenas de anos mais velho do que se pensava. Análises genéticas ainda revelaram que ele tinha um ancestral neandertal, distante de quatro a seis gerações.
“Isso significa que, quanto mais antigo o Oase 1 é, mais neandertais andavam pela Europa”, explica Higham. Em contrapartida, o adolescente cujos fósseis foram desenterrados na Sibéria viveu mil anos à frente do que se pensava e isso, segundo o cronólogo, muda a história da chegada dos humanos modernos na Sibéria.a













{kind=link}
{kind=link}
{kind=link}
{kind=link}