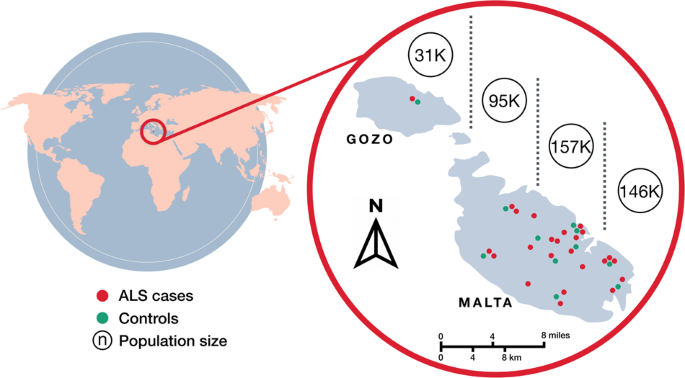
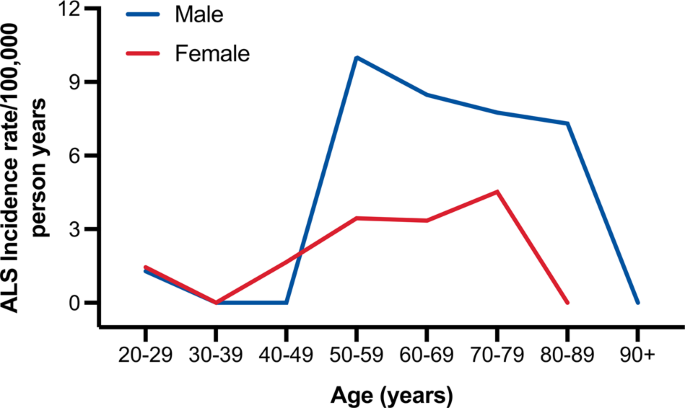
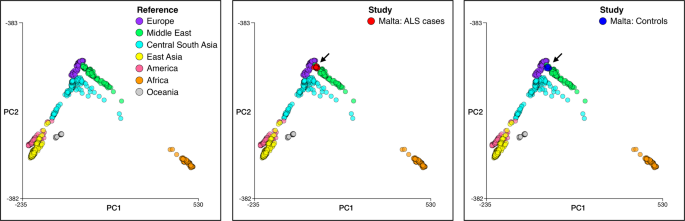
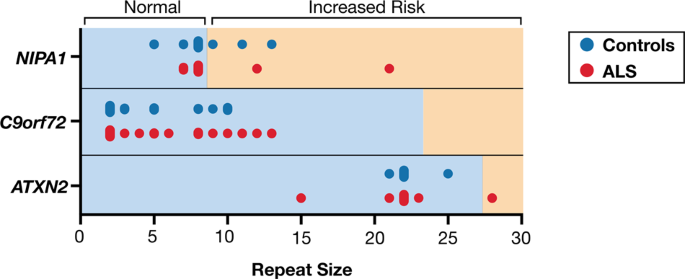
- 1
Brown RH, Al-Chalabi A. Esclerose lateral amiotrófica. N Engl J Med. 2017; 377: 162–72.
CAS PubMed Google Scholar
- 2
van Es MA, Hardiman O, Chio A, Al-Chalabi A, Pasterkamp RJ, Veldink JH, et al. Esclerose lateral amiotrófica. Lanceta. 2017; 390: 2084–98.
PubMed Google Scholar
- 3
Chio A, Logroscino G, Traynor BJ, Collins J, Simeone JC, Goldstein LA, et al. Epidemiologia global da esclerose lateral amiotrófica: uma revisão sistemática da literatura publicada. Neuroepidemiologia. 2013; 41: 118–30.
CAS PubMed PubMed Central Google Scholar
- 4
Byrne S, Walsh C, Lynch C, Bede P, Elamin M, Kenna K, et al. Taxa de esclerose lateral amiotrófica familiar: uma revisão sistemática e meta-análise. J Neurol Neurosurg Psychiatry. 2011; 82: 623–7.
PubMed Google Scholar
- 5
Al-Chalabi A, Fang F, Hanby MF, Leigh PN, Shaw CE, Ye W, et al. Uma estimativa da herdabilidade da esclerose lateral amiotrófica usando dados de gêmeos. J Neurol Neurosurg Psychiatry. 2010; 81: 1324–6.
CAS PubMed PubMed Central Google Scholar
- 6
Ryan M, Heverin M, McLaughlin RL, Hardiman O. Risco ao longo da vida e herdabilidade da esclerose lateral amiotrófica. JAMA Neurol. 2019; 76: 1367–74.
PubMed Central Google Scholar
- 7
Trabjerg BB, Garton FC, van Rheenen W., Fang F, Henderson RD, Mortensen PB, et al. ALS em registros dinamarqueses: herdabilidade e links para transtornos psiquiátricos e cardiovasculares. Neurol Genet. 2020; 6: e398.
PubMed PubMed Central Google Scholar
- 8
Zou ZY, Zhou ZR, Che CH, Liu CY, He RL, Huang HP. Epidemiologia genética da esclerose lateral amiotrófica: uma revisão sistemática e meta-análise. J Neurol Neurosurg Psychiatry. 2017; 88: 540–9.
PubMed Google Scholar
- 9
Gibson SB, Downie JM, Tsetsou S, Feusier JE, Figueroa KP, Bromberg MB, et al. O risco genético em evolução para ELA esporádica. Neurologia. 2017; 89: 226–33.
CAS PubMed PubMed Central Google Scholar
- 10
Kenna KP, McLaughlin RL, Byrne S, Elamin M, Heverin M, Kenny EM, et al. Delineando a heterogeneidade genética de ALS usando sequenciamento de alto rendimento direcionado. J Med Genet. 2013; 50: 776–83.
CAS PubMed PubMed Central Google Scholar
- 11
Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, et al. As expansões de poliglutamina de comprimento intermediário da Ataxina-2 estão associadas a um risco aumentado de ELA. Natureza. 2010; 466: 1069–75.
CAS PubMed PubMed Central Google Scholar
- 12
van Es MA, Veldink JH, Saris CG, Blauw HM, van Vught PW, Birve A, et al. O
estudo de associação de todo o genoma identifica 19p13.3 (UNC13A) e
9p21.2 como loci de suscetibilidade para esclerose lateral amiotrófica
esporádica. Nat Genet. 2009; 41: 1083–7.
PubMed Google Scholar
- 13
van Rheenen W., Shatunov A., Dekker AM, McLaughlin RL, Diekstra FP, Pulit SL, et al. As
análises de associação de todo o genoma identificam novas variantes de
risco e a arquitetura genética da esclerose lateral amiotrófica. Nat Genet. 2016; 48: 1043–8.
PubMed PubMed Central Google Scholar
- 14
Taylor JP, Brown RH Jr., Cleveland DW. Decodificando ALS: dos genes ao mecanismo. Natureza. 2016; 539: 197–206.
PubMed PubMed Central Google Scholar
- 15
Ly CV, Miller TM. Novos oligonucleotídeos antisense e terapias virais para esclerose lateral amiotrófica. Curr Opin Neurol. 2018; 31: 648–54.
CAS PubMed PubMed Central Google Scholar
- 16
Arcos-Burgos M, Muenke M. Genética de isolados de população. Clin Genet. 2002; 61: 233–47.
CAS PubMed Google Scholar
- 17
Caruana J. Population genetics of Western Mediterranean Islands — Malta: a case study. Manchester: Universidade de Manchester; 2012
Google Scholar
- 18
Capelli C, Redhead N, Romano V, Cali F, Lefranc G, Delague V, et al. Estrutura populacional na bacia do Mediterrâneo: uma perspectiva do cromossomo Y. Ann Hum Genet. 2006; 70: 207–25.
CAS PubMed Google Scholar
- 19
Cassar M, Farrugia C, Vidal C. Frequências alélicas de 14 loci STR na população de Malta. Leg Med. 2008; 10: 153–6.
CAS Google Scholar
- 20
Borg J, Papadopoulos P, Georgitsi M, Gutierrez L, Grech G, Fanis P, et al. A haploinsuficiência para o fator de transcrição eritróide KLF1 causa persistência hereditária da hemoglobina fetal. Nat Genet. 2010; 42: 801–5.
CAS PubMed PubMed Central Google Scholar
- 21
Dito E, Chong JX, Hempel M, Denecke J, Soler P, Strom T, et al. A sobrevivência além do período perinatal expande os fenótipos causados por mutações em GLE1. Am J Med Genet A. 2017; 173: 3098–103.
CAS PubMed PubMed Central Google Scholar
- 22
Brooks BR, Miller RG, Swash M, Munsat TL. Grupo
de Pesquisa da Federação Mundial de Neurologia em Neurônio Motor D. El
Escorial revisitado: critérios revisados para o diagnóstico de
esclerose lateral amiotrófica. Amyotroph Lateral Scler Outro Mot Neuron Disord. 2000; 1: 293–9.
CAS Google Scholar
- 23
Ludolph A, Drory V, Hardiman O, Nakano I, Ravits J, Robberecht W, et al. Uma revisão dos critérios do El Escorial - 2015. Amyotroph Lateral Scler Frontotemporal Degener. 2015; 16: 291–2.
PubMed Google Scholar
- 24
Dolzhenko E, van Vugt J, Shaw RJ, Bekritsky MA, van Blitterswijk M, Narzisi G, et al. Detecção de expansões repetidas longas a partir de dados de sequência do genoma completo sem PCR. Genome Res. 2017; 27: 1895–903.
CAS PubMed PubMed Central Google Scholar
- 25
Seelow
D, Schuelke M. HomozygosityMapper2012-colmatando a lacuna entre o
mapeamento de homozigosidade e o sequenciamento profundo. Nucleic Acids Res. 2012; 40: W516–20.
CAS PubMed PubMed Central Google Scholar
- 26
Nicolas A, Kenna KP, Renton AE, Ticozzi N, Faghri F, Chia R, et al. As análises de todo o genoma identificam KIF5A como um novo gene ALS. Neuron. 2018; 97: 1268–83 e6.
CAS PubMed PubMed Central Google Scholar
- 27
van der Spek RAA, van Rheenen W., Pulit SL, Kenna KP, van den Berg LH, Veldink JH, et al. O projeto MinE databrowser: trazendo o sequenciamento de todo o genoma em larga escala em ALS para pesquisadores e o público. Amyotroph Lateral Scler Frontotemporal Degener. 2019; 20: 432–40.
PubMed Google Scholar
- 28
Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, et al. Comparação
e integração de métodos de predição de deletérios para SNVs não
sinônimos em estudos de sequenciamento de exoma completo. Hum Mol Genet. 2015; 24: 2125–37.
CAS PubMed Google Scholar
- 29
Byrne S, Heverin M, Elamin M, Bede P, Lynch C, Kenna K, et al. Agregação
de doenças neurológicas e neuropsiquiátricas em famílias com esclerose
lateral amiotrófica: um estudo de coorte caso-controle de base
populacional de esclerose lateral amiotrófica familiar e esporádica. Ann Neurol. 2013; 74: 699–708.
PubMed Google Scholar
- 30
O'Brien M., Burke T., Heverin M., Vajda A., McLaughlin R., Gibbons J, et al. Agrupamento
de doenças neuropsiquiátricas em parentes de primeiro e segundo grau de
pacientes com esclerose lateral amiotrófica. JAMA Neurol. 2017; 74: 1425–30.
PubMed PubMed Central Google Scholar
- 31
Longinetti E, Mariosa D, Larsson H, Ye W, Ingre C, Almqvist C, et al. Doenças neurodegenerativas e psiquiátricas em famílias com esclerose lateral amiotrófica. Neurology 2017; 89: 578–85.
PubMed PubMed Central Google Scholar
- 32
Peters S, Visser AE, D'Ovidio F, Vlaanderen J, Portengen L, Beghi E, et al. Modificação
do efeito da associação entre o tabagismo total e risco de ALS por
intensidade, duração e tempo desde o abandono: Euro-MOTOR. J Neurol Neurosurg Psychiatry. 2020; 91: 33–9.
PubMed Google Scholar
- 33
Visser AE, Rooney JPK, D'Ovidio F, Westeneng HJ, Vermeulen RCH, Beghi E, et al. Estudo
multicêntrico, transcultural, de base populacional e caso-controle da
atividade física como fator de risco para esclerose lateral amiotrófica.
J Neurol Neurosurg Psychiatry. 2018; 89: 797–803.
PubMed Google Scholar
- 34
Di Gaetano C, Voglino F, Guarrera S, Fiorito G, Rosa F, Di Blasio AM, et al. Uma visão geral da estrutura genética dentro da população italiana a partir de dados de todo o genoma. PLoS ONE. 2012; 7: e43759.
PubMed PubMed Central Google Scholar
- 35
Sarno S, Boattini A, Pagani L, Sazzini M, De Fanti S, Quagliariello A, et al. Camadas
de mistura antigas e recentes na Sicília e no sul da Itália traçam
múltiplas rotas de migração ao longo do Mediterrâneo. Sci Rep. 2017; 7: 1984.
PubMed PubMed Central Google Scholar
- 36
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. Uma expansão de repetição de hexanucleotídeo em C9ORF72 é a causa do ALS-FTD ligado ao cromossomo 9p21. Neuron. 2011; 72: 257–68.
CAS PubMed PubMed Central Google Scholar
- 37
Smith BN, Newhouse S, Shatunov A, Vance C, Topp S, Johnson L, et al. A mutação de expansão C9ORF72 é uma causa comum de ALS +/- FTD na Europa e tem um único fundador. Eur J Hum Genet. 2013; 21: 102–8.
CAS PubMed Google Scholar
- 38
Blauw HM, van Rheenen W., Koppers M., Van Damme P., Waibel S, Lemmens R., et al. As expansões de repetição de polialanina NIPA1 estão associadas à esclerose lateral amiotrófica. Hum Mol Genet. 2012; 21: 2497–502.
CAS PubMed Google Scholar
- 39
Van Damme P, Veldink JH, van Blitterswijk M, Corveleyn A, van Vught PW, Thijs V, et al. O tamanho de repetição ATXN2 CAG expandido em ALS identifica a sobreposição genética entre ALS e SCA2. Neurologia. 2011; 76: 2066–72.
PubMed Google Scholar
- 40
Tazelaar GHP, Dekker AM, van Vugt J, van der Spek RA, Westeneng HJ, Kool L, et al. Associação de expansões de repetição NIPA1 com esclerose lateral amiotrófica em uma grande coorte internacional. Neurobiol Aging. 2019; 74: 234 e9–15.
Google Scholar
- 41
Ragonese P, Cellura E, Aridon P, D'Amelio M, Spataro R, Taiello AC, et al. Incidência de esclerose lateral amiotrófica na Sicília: um estudo de base populacional. Amyotroph Lateral Scler. 2012; 13: 284–7.
PubMed Google Scholar
- 42
Logroscino G, Beghi E, Zoccolella S, Palagano R, Fraddosio A, Simone IL, et al. Incidência de esclerose lateral amiotrófica no sul da Itália: um estudo de base populacional. J Neurol Neurosurg Psychiatry. 2005; 76: 1094–8.
CAS PubMed PubMed Central Google Scholar
- 43
Zoccolella S, Beghi E, Palagano G, Fraddosio A, Samarelli V, Lamberti P, et al. Sinais e sintomas no diagnóstico de esclerose lateral amiotrófica: um estudo de base populacional no sul da Itália. Eur J Neurol. 2006; 13: 789–92.
CAS PubMed Google Scholar
- 44
Kacem I, Sghaier I, Bougatef S, Nasri A, Gargouri A, Ajroud-Driss S, et al. Aspectos epidemiológicos e clínicos da esclerose lateral amiotrófica em uma coorte tunisiana. Amyotroph Lateral Scler Frontotemporal Degener. 2020; 21: 131–9.
CAS PubMed Google Scholar
- 45
Demetriou CA, Hadjivasiliou PM, Kleopa KA, Christou YP, Leonidou E, Kyriakides T, et al. Epidemiologia da esclerose lateral amiotrófica na República de Chipre: um estudo retrospectivo de 25 anos. Neuroepidemiologia. 2017; 48: 79–85.
PubMed Google Scholar
- 46
Logroscino G, Traynor BJ, Hardiman O, Chio A, Mitchell D, Swingler RJ, et al. Incidência de esclerose lateral amiotrófica na Europa. J Neurol Neurosurg Psychiatry. 2010; 81: 385–90.
PubMed Google Scholar
- 47
Marin B, Boumediene F, Logroscino G, Couratier P, Babron MC, Leutenegger AL, et al. Variação na incidência mundial de esclerose lateral amiotrófica: uma meta-análise. Int J Epidemiol. 2017; 46: 57–74.
PubMed Google Scholar
- 48
Pradas J, Puig T, Rojas-Garcia R, Viguera ML, Gich I, Logroscino G, et al. Esclerose lateral amiotrófica na Catalunha: um estudo de base populacional. Amyotroph Lateral Scler Frontotemporal Degener. 2013; 14: 278–83.
PubMed Google Scholar
- 49.
Mandrioli J, Biguzzi S, Guidi C, Venturini E, Sette E, Terlizzi E, et al. Epidemiologia da esclerose lateral amiotrófica na região de Emilia Romagna (Itália): um estudo de base populacional. Amyotroph Lateral Scler Frontotemporal Degener. 2014; 15: 262–8.
PubMed Google Scholar
- 50
Scialo C, Novi G, Bandettini di Poggio M, Canosa A, Sormani MP, Mandich P. et al. Epidemiologia clínica da esclerose lateral amiotrófica na Ligúria, Itália: uma atualização do registro LIGALS. Amyotroph Lateral Scler Frontotemporal Degener. 2016; 17: 535–42.
PubMed Google Scholar
- 51
Palese F, Sartori A, Verriello L, Ros S, Passadore P, Manganotti P, et al. Epidemiologia
da esclerose lateral amiotrófica em Friuli-Venezia Giulia, Nordeste da
Itália, 2002-2014: um estudo retrospectivo de base populacional. Amyotroph Lateral Scler Frontotemporal Degener. 2019; 20: 90–9.
PubMed Google Scholar
- 52
Borghero G, Pugliatti M, Marrosu F, Marrosu MG, Murru MR, Floris G, et al. Arquitetura genética de ALS na Sardenha. Neurobiol Aging. 2014; 35: 2882 e7–12.
Google Scholar
- 53
Ryan M., Heverin M., Doherty MA, Davis N., Corr EM, Vajda A, et al. Determinando a incidência de familialidade em ALS: Um estudo de tendências temporais na Irlanda de 1994 a 2016. Neurol Genet. 2018; 4: e239.
PubMed PubMed Central Google Scholar
- 54
McLaughlin RL, Schijven D, van Rheenen W., van Eijk KR, O'Brien M, Kahn RS, et al. Correlação genética entre esclerose lateral amiotrófica e esquizofrenia. Nat Commun. 2017; 8: 14774.
CAS PubMed PubMed Central Google Scholar
- 55
Hadano S, Hand CK, Osuga H, Yanagisawa Y, Otomo A, Devon RS, et al. Um gene que codifica um regulador GTPase putativo sofre mutação na esclerose lateral amiotrófica familiar 2. Nat Genet. 2001; 29: 166–73.
CAS PubMed Google Scholar
- 56
Hentati A, Bejaoui K, Pericak-Vance MA, Hentati F, Speer MC, Hung WY, et al. Ligação da esclerose lateral amiotrófica familiar recessiva ao cromossomo 2q33-q35. Nat Genet. 1994; 7: 425–8.
CAS PubMed Google Scholar
- 57
Daoud H, Zhou S, Noreau A, Sabbagh M, Belzil V, Dionne-Laporte A, et al. O sequenciamento do exoma revela mutações SPG11 causando ALS juvenil. Neurobiol Aging. 2012; 33: 839 e5–9.
Google Scholar
- 58
Orlacchio A, Babalini C, Borreca A, Patrono C, Massa R, Basaran S, et al. Mutações no SPATACSIN causam esclerose lateral amiotrófica juvenil autossômica recessiva. Cérebro. 2010; 133: 591–8.
PubMed PubMed Central Google Scholar
- 59.
van Blitterswijk M, van Es MA, Hennekam EA, Dooijes D, van Rheenen W, Medic J, et al. Evidência de uma base oligogênica da esclerose lateral amiotrófica. Hum Mol Genet. 2012; 21: 3776–84.
PubMed Google Scholar
- 60
Mitchell J, Paul P, Chen HJ, Morris A, Payling M, Falchi M, et al. A esclerose lateral amiotrófica familiar está associada a uma mutação na D-aminoácido oxidase. Proc Natl Acad Sei USA. 2010; 107: 7556–61.
CAS PubMed Google Scholar
- 61
Takahashi Y, Fukuda Y, Yoshimura J, Toyoda A, Kurppa K, Moritoyo H, et al. Mutações ERBB4 que interrompem a via neuregulina-ErbB4 causam esclerose lateral amiotrófica tipo 19. Am J Hum Genet. 2013; 93: 900–5.
CAS PubMed PubMed Central Google Scholar
- 62
Chen YZ, Bennett CL, Huynh HM, Blair IP, Puls I, Irobi J, et al. Mutações do gene da helicase DNA / RNA em uma forma de esclerose lateral amiotrófica juvenil (ALS4). Am J Hum Genet. 2004; 74: 1128–35.
CAS PubMed PubMed Central Google Scholar
- 63
Munch C, Sedlmeier R, Meyer T, Homberg V, Sperfeld AD, Kurt A, et al. Mutações pontuais da subunidade p150 do gene da dinactina (DCTN1) em ALS. Neurologia. 2004; 63: 724–6.
CAS PubMed Google Scholar
- 64
Tripolszki K, Torok D, Goudenege D, Farkas K, Sulak A, Torok N, et al. O sequenciamento de alto rendimento revelou uma nova mutação SETX em um paciente húngaro com esclerose lateral amiotrófica. Brain Behav. 2017; 7: e00669.
PubMed PubMed Central Google Scholar
- 65
Aquilina B, Cauchi RJ. Modelagem de doenças do neurônio motor em moscas-das-frutas: lições da atrofia muscular espinhal. J Neurosci Methods. 2018; 310: 3–11.
CAS PubMed Google Scholar

:max_bytes(150000):strip_icc():format(webp)/H1N1_virus-56a09b633df78cafdaa32fa0.jpg)