Inclusão e ética
Os
estudos genéticos de doenças humanas antigas esclarecem como as
populações do passado prosperaram e lidaram com problemas de saúde, o
que pode desencadear preocupações como a estigmatização devido a doenças
ou direitos e questões legais entre as pessoas que vivem hoje. As
injustiças históricas, a colonização e a expropriação complicaram
frequentemente a capacidade das comunidades indígenas de afirmarem e
manterem os seus direitos territoriais num quadro jurídico ou
administrativo. É, portanto, crucial considerar, além dos aspectos
científicos, também as perspectivas das comunidades e pessoas vivas
(indígenas) ao realizar este trabalho. 55 .
Aqui
estudamos restos humanos de indivíduos totalmente anônimos que morreram
há mais de 1.000 anos e foram sepultados no sítio arqueológico
Jabuticabeira II, no município de Tubarão, no estado de Santa Catarina,
Brasil. Este local foi escavado por P. de Blasis e equipe 56 ,
financiado pela Fundação de Amparo à Pesquisa do Estado de São Paulo
(FAPESP). e foi obtida autorização de pesquisa junto ao Instituto do
Patrimônio Histórico e Artístico Nacional (IPHAN), conforme
correspondência 1793/2019 GAB PRESI-IPHAN do processo
01506.000720/2019-65 de K. Santos Bogia. A utilização das amostras dos
restos mortais para este estudo também foi aprovada por P. de Blasis,
guardião das coleções Jabuticabeira II do Museu de Arqueologia e
Etnologia da Universidade de São Paulo. Os restos mortais foram
curados, estudados e amostrados por SE e equipe da Universidade de São
Paulo até 2016 e, posteriormente, no Museu de História Natural de Viena.
Os territórios e sítios que abrangem o Rio Grande do Sul, Santa
Catarina e Paraná são inerentes à herança ancestral das comunidades
Kaingang, Guarani e Xokleng (também chamados de 'povo Sol' ou 'grupo
costeiro'), que ainda vivem hoje na região. Estas sociedades não só
utilizaram a região para mobilização e migração em busca de
abastecimento alimentar, mas também percorreram tradicionalmente
distâncias significativas, deixando um rasto de marcas culturais,
particularmente no domínio das práticas funerárias. Em um estudo
anterior 57,
samples of five of the individuals exhumed at Jabuticabeira II were
studied, revealing some genetic affinity with the Kaingang (a
Ge-speaking group of Southern Brazil). However, to the best of our
knowledge, the Kaingang are not seen as direct descendants of sambaqui
societies, nor do they identify with the people who once dwelled at
Jabuticabeira II or request their remains. Finally, research in the
Instituto Socioambiental (https://www.socioambiental.org/;
for the defence of Brazilian socio-environmental diversity, including
Indigenous Rights) states that the region around Jabuticabeira II is not
part of any Indigenous reserve, nor are there claims of groups for
territorial rights of this region or for the archaeological remains of
this site (P. de Blasis, personal communication).
Os processos
degenerativos, muitas vezes resultantes de contextos de marginalização,
conflito e deslocamento, testemunham o impacto das relações históricas
dos grupos indígenas com os colonizadores e invasores. As aflições e
doenças vividas por estes grupos carregam ramificações históricas e
ambientais de notável significado, garantindo reconhecimento e exame
explícitos. Em relação à possível estigmatização das comunidades locais
(indígenas) e das pessoas afetadas pelo bejel, deve-se ressaltar que
esta doença contagiosa é uma doença endêmica, principalmente não
sexualmente transmissível, comum em regiões quentes onde as pessoas
vivem em contato próximo umas com as outras, não têm necessidade de
cobrir especialmente roupas e compartilhar utensílios. Hoje, o bejel,
que pode levar à estigmatização devido a feridas desfigurantes, ocorre
especialmente em comunidades do Mediterrâneo Oriental e da África
Ocidental com acesso limitado a cuidados médicos modernos. Embora a
Organização Mundial da Saúde perceba a importância das ações tomadas
para erradicar o bejel em todo o mundo desde 1949 (WHA2.36 Bejel e
outras treponematoses ( https://www.who.int/publications/i/item/wha2.36) ), a doença não é vista como um problema atual de saúde pública no Brasil, como é para alguns outros países 58 , 59 .
Isto contrasta com as altas prevalências de doenças sexualmente
transmissíveis, como o HIV e a sífilis venérea, que afetam as
comunidades indígenas no Brasil (Em São Paulo, ação em aldeias promove
debate e testagem rápida de HIV e Sífilis — Fundação Nacional dos Povos
Indígenas (
http://www.gov.br/funai/pt-br/assuntos/noticias/2019/em-sao-paulo-acao-em-aldeias-promove-debate-e-testagem-rapida-de-hiv-e-
sífilis )). É notável, entretanto, que, no contexto
arqueológico, nada implicava que aqueles povos pré-históricos de
Jabuticabeira II portadores da treponematose local teriam sido
discriminados em sua época e cultura.
Além disso, as descrições
culturalmente insensíveis em artigos de investigação paleogenómica são
uma questão ética de preocupação 60 .
Para garantir a discrição, selecionamos expressões potencialmente
insensíveis ou discriminatórias no manuscrito. É importante ressaltar
que tivemos a ajuda inestimável de E. Krenak, líder da Cultural Survival
no Brasil, ativista indígena e doutorando na Universidade de Viena,
para analisar criticamente nossos textos e fornecer conselhos sobre o
uso eticamente correto e justo da terminologia.
Informação arqueológica
Os sambaquis da região de Laguna
Um
sambaqui é o tipo predominante de sítio arqueológico na costa
brasileira: um monturo ou montículo de conchas construído pelo homem, de
dimensões variadas, localizado em áreas ricas em recursos, como lagoas,
manguezais ou estuários. Sambaquis consistem em sedimentos
inorgânicos, conchas de moluscos, restos de alimentos e matéria orgânica
misturados em intrincadas estratigrafias associadas a funções
domésticas e/ou funerárias. 61 .
Mais de 1.000 sambaquis estão mapeados ao longo dos 7.500 km de
extensão da costa brasileira e são datados entre 7.500 e 1.000 anos bp 61 , 62 .
Pesquisas arqueológicas recentes sugerem que essas populações
construtoras de sambaquis eram sedentárias, com uma abundante e estável
subsistência marinha, horticultura e alto crescimento populacional. 61 , 63 , rituais funerários elaborados 64 e apropriação da paisagem 56 .
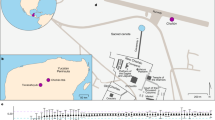
Jabuticabeira II excavation site
Jabuticabeira
II (UTM 22 J − 0699479E; 6835488 S) é um sambaqui de médio porte (400 ×
250 × 10 m de altura), assentado sobre uma paleoduna e localizado na
região de Laguna, área de maior densidade de sambaquis do sul Costa
brasileira, a 3 km da Laguna do Camacho, uma das diversas fontes de água
associadas a um sistema geológico barreira-lagoa formado durante o
Holoceno (Fig. 1
). Jabuticabeira II, construída ao longo de quase mil anos, é um dos
65 sambaquis mapeados ao redor do sistema lagunar. Este grande número
de assentamentos e sua história de ocupação cronologicamente sobreposta
atestam uma ocupação bastante densa e intensas interações dos
construtores de sambaqui entre 7.500 e 900 anos calibrados (ano cal) bp 56.
According to stratigraphic studies, Jabuticabeira II is the result of
incremental funerary rituals accumulated over centuries. Although
Jabuticabeira II was not completely excavated, 204 burials containing
the remains of 282 individuals were exhumed from a 373 m2 area64. Radiocarbon dates of Jabuticabeira II stratigraphy56,64 suggest a long occupation period between 1214–830 cal bc and 118–413 cal ad or 3137–2794 to 1860–1524 cal yr bp (2σ),
roughly in line with the radiocarbon datings from bone material of the
four individuals in this study, ranging from 350 cal bc to 573 cal ad.
Os
restos mortais do sambaqui Jabuticabeira II foram encontrados em
sepultamentos simples, duplos e múltiplos, dispersos em aglomerados. Os
esqueletos recuperados estavam em sua maioria incompletos, evitando
estimativas categóricas de idade e sexo ou outros achados osteológicos.
O padrão de sepultamento era bem flexionado e sugeria tratamento
intencional do corpo antes do internamento. O pequeno tamanho das
sepulturas sugeria que os corpos sofreram dessecação ou decomposição
prévia de tecidos moles, mas não o suficiente para produzir
desarticulação completa (os ossos das mãos e dos pés foram encontrados
articulados). Muitos enterros vêm de perfis e estão incompletos. Os
ossos de vários indivíduos estão manchados de ocre vermelho 65 , uma prática comum em sítios arqueológicos do estado de Santa Catarina 66 , 67 .
As oferendas são comuns em contextos funerários de sambaqui e incluem
adornos confeccionados com material faunístico e ferramentas líticas em
diversas formas, desde entulhos até ferramentas polidas e zoólitos, com
diferenças na frequência de ocorrência entre diferentes locais e
estratos 68 . A oferta mais comum na Jabuticabeira II era o peixe.
Ao
todo, 99 indivíduos de Jabuticabeira II, com e sem alterações ósseas
sugestivas de infecção, foram triados quanto ao conteúdo de DNA do
patógeno. 37 amostras foram consideradas positivas para DNA
treponêmico na triagem inicial e quatro amostras produziram dados
suficientes para a reconstrução do genoma de T. pallidum (Tabela Suplementar 1 ).
Palaeopathological analysis of treponematoses
Bioarchaeological
analyses showed results compatible with increasing population growth
and high population density in Jabuticabeira II, including high
frequencies of nonspecific stress markers69 and occasional infant stress70, but no evidence of trauma associated with interpersonal conflicts over resources or territory69.
Há, no entanto, evidências de doenças sistêmicas transmissíveis em Jabuticabeira II e outros sambaquis locais brasileiros 30. Eleven 14C
accelerator mass spectrometry dates obtained directly from the
presumably treponematosis-affected individuals suggest that these
diseases are very old on the east coast of South America, with a
time-range between 6,300 and 500 yr BP. Among the possible treponemal
cases based on osteological analysis, three came from Jabuticabeira II.
However, these did not overlap with the individuals yielding the
detected genetic evidence in this study.
Informações sobre indivíduos
Indivíduo 41A-L2.05-E4, amostra ZH1390
O indivíduo é um homem adulto de constituição robusta, com estatura estimada em 150,49 ± 2,6 cm (ref. 70 ).
Embora fragmentados, os ossos deste indivíduo constituíam um esqueleto
quase completo (80%), articulado e enterrado em uma matriz oval rica em
conchas em posição hiperflexionada. Os ossos do indivíduo apresentavam
sinais de doença infecciosa sistêmica em membros inferiores. Fêmures,
tíbias e fíbulas apresentam periostite generalizada discreta e
osteoartrose. Também foi observado alargamento na porção lateral das
clavículas. Segundo Filippini et al. 30 , aplicando o método ESPIRAL 71 ,
a doença desse indivíduo poderia ser classificada de forma não
conclusiva como sífilis, bouba ou bejel. A amostragem foi realizada em
lesão ativa no fragmento da tíbia.
FS9–L3–T2 individual, amostra ZH1540
A amostra vem de um conjunto de ossos misturados,
provavelmente de mais de um indivíduo. Os ossos atribuídos a este
indivíduo consistem em vários elementos esqueléticos, alguns com
alterações patológicas, como osteomielite grave no terço distal do úmero
direito, periostite grave na ulna esquerda, periostite na diáfise da
fíbula e dois corpos vertebrais com osteofitose. A amostra foi retirada
do fragmento da fíbula, na área com periostite.
FS3B-L3-T4 individual, amostra ZH1541
A amostra vem de um de três indivíduos separados,
encontrados misturados. Os elementos esqueléticos pertencentes a este
adulto robusto de idade e sexo desconhecidos incluem um rádio esquerdo
com artrite, um fragmento da ulna esquerda (muito robusto), um fragmento
do úmero esquerdo, fragmentos de um fêmur, uma tíbia e uma fíbula e um
primeiro metatarso. A amostra foi retirada de um fragmento de fêmur,
sob a superfície imediata do osso, para melhor evitar a possível
introdução de contaminantes externos.
Individual 2B-L6-E3, ZH1557
A amostra provém de um indivíduo provavelmente
adulto do sexo masculino. O indivíduo estava articulado e em posição
flexionada com outro indivíduo adulto do sexo feminino enterrado em
cima. Os achados osteopatológicos nos ossos do indivíduo amostrado
incluíram sinais de doença articular degenerativa, osteoartrite
intervertebral lombar grave, escoliose e possíveis lesões na patela. No
entanto, não foram observadas lesões típicas sugestivas de infecção
treponêmica. A amostra foi retirada de um pequeno pedaço de osso longo,
sob a superfície imediata do osso, para melhor evitar a possível
introdução de contaminantes externos.
Correção do efeito do reservatório marinho para 14 C namoro
A
datação por radiocarbono foi realizada pelo Laboratório de Física de
Feixes de Íons da ETH Zurich (laboratório número: ETH-127328) utilizando
colágeno ósseo purificado por um método de ultrafiltração modificado 72 .
A calibração dos dados foi feita com OxCal v4.4.4. A dieta dos
habitantes de Jabuticabeira II, constituída substancialmente por fontes
alimentares marinhas, produz efeito reservatório nas datas de
radiocarbono calculadas como idade média de 247,8 ( σ = 103,7) anos 73 .
Considerando a alta contribuição de carbono marinho para o colágeno
ósseo dos indivíduos de Jabuticabeira II, as datas de radiocarbono foram
modeladas com Calib Rev 8.20 74 ( http://calib.org/calib/calib.htm ) usando a curva de calibração Mixed Marine SHCal20 75 , 76 e aplicando o valor médio estimado de correção de reservatórios de radiocarbono marinhos locais (Δ R ) de -126 ± 29 para a costa sul do Brasil (banco de dados de Correção de Reservatórios Marinhos) 73,77.
We considered the average relative contribution of marine carbon to
collagen derived from Bayesian Mixing Models for Jabuticabeira II
individuals, calculated at a mean value of 42.5%78,79. Para as estimativas individuais para as amostras, consulte a Tabela Suplementar 2 .
Sample processing
Samples
were documented and carried through sampling, DNA extraction, library
preparation and library indexing in facilities dedicated to ancient DNA
work at the University of Zurich, including decontamination of samples,
laboratory equipment and reagents with UV irradiation and using
protective clothing and minimum contamination-risk working methods.
Todas
as etapas pós-amplificação foram realizadas nas instalações
laboratoriais regulares disponíveis para o Grupo de Paleogenética do
Instituto de Medicina Evolutiva (IEM) da Universidade de Zurique (UZH).
O sequenciamento de DNA foi realizado nas instalações de Sequenciamento
de Próxima Geração das Instalações Principais do BioCenter de Viena
(VBCF) ou no Centro de Genômica Funcional da Universidade de Zurique
(FGCZ).
Extração de DNA antigo
Todas
as superfícies das amostras foram irradiadas com luz ultravioleta para
minimizar a contaminação potencial do DNA moderno. O pó ósseo foi
obtido utilizando-se broca odontológica e brocas com cabeça diamantada.
A extração de DNA foi realizada em cerca de 50-100 mg de pó de osso, de
acordo com um protocolo de extração bem estabelecido para DNA antigo. 80 .
Os controles negativos para processos de extração e biblioteca foram
processados paralelamente em todos os experimentos, um controle para
cada dez amostras, sequenciados e comparados bioinformaticamente com
seus correspondentes lotes de amostras, como precaução contra possível
contaminação.
Preparação da biblioteca
Bibliotecas
de DNA de cadeia dupla foram produzidas para triagem inicial com
sequenciamento shotgun, sem tratamento UDG (ou seja, tratamento químico
com o objetivo de limitar os danos relacionados à idade no DNA). Duas
bibliotecas adicionais para cada uma das amostras potencialmente
positivas da primeira rodada de captura foram produzidas para maximizar a
complexidade do DNA. Para a preparação de bibliotecas de DNA, 20 µl de
extrato de DNA foram convertidos em bibliotecas de DNA de fita dupla 31 .
Códigos de barras (índices) específicos da amostra foram adicionados a
ambas as extremidades dos fragmentos de DNA nas bibliotecas 81 . As bibliotecas indexadas foram então amplificadas para atingir uma concentração mínima de DNA de aproximadamente 90 ng ml −1 . A amplificação foi realizada utilizando 1 × tampão Herculase II, 0,4 mM IS5 e 0,4 mM IS6 primer 81 ,
DNA polimerase de fusão Herculase II (Agilent Technologies), dNTPs 0,25
mM (100 mM; 25 mM cada dNTP) e 5 ml de biblioteca indexada como modelo
de DNA. Foram preparadas quatro reações por biblioteca e o volume total
da reação de amplificação foi de 100 ml. O perfil térmico incluiu uma
desnaturação inicial por 2 min a 95 °C e 3-18 ciclos, dependendo da
concentração de DNA após a indexação das bibliotecas, desnaturação por
30 s a 95 °C, 30 s de recozimento a 60 °C e 30 s de recozimento a 60 °C.
alongamento a 72°C, seguido por uma etapa de alongamento final por 5
min a 72°C. Todas as divisões de uma biblioteca indexada foram reunidas
e purificadas utilizando o kit de purificação QIAGEN MinElute PCR. As
bibliotecas de DNA foram então quantificadas com D1000 ScreenTape em um
Agilent 2200 TapeStation (Agilent Technologies) e combinadas em pools
equimolares para sequenciamento.
Pathogen screening
Os dados do Shotgun foram usados para uma triagem inicial das 99 amostras candidatas, com o software Kraken2 82 , e 41 amostras que tiveram mais de 7 ocorrências para T. pallidum
foram selecionadas para enriquecimento alvo. As amostras selecionadas
foram submetidas a um processo de enriquecimento alvo e posteriormente
processadas pelo FastQ Screen v0.15.1 83 para verificar o número de leituras mapeadas em relação a três genomas de referência representativos de alta qualidade da subespécie de T. pallidum (CDC2, BosniaA e Nichols). As nove amostras mais promissoras (> 5.000 acessos de Kraken a T. pallidum
após a primeira rodada de captura em solução) foram transformadas em
duas bibliotecas extras e recapturadas conforme explicado em detalhes
nas seções a seguir.
Enriquecimento alvo para T. pallidum DNA de
O
enriquecimento de bibliotecas de cadeia dupla em todo o genoma foi
realizado através de kits de enriquecimento de alvo personalizados
(Arbor Bioscience). Iscas de RNA com comprimento de 60 nucleotídeos e
densidade de ladrilhos de 4 pb foram projetadas com base em três genomas
de referência: Nichols ( CP004010.2 ), SS14 ( CP000805.1 ), Fribourg-Blanc ( CP003902
). Conjuntos de bibliotecas de 500 ng foram enriquecidos de acordo
com as instruções do fabricante. As bibliotecas capturadas foram
amplificadas em reações de 100 µl contendo 1 unidade de DNA polimerase
de fusão Herculase II (Agilent), 1 × tampão de reação Herculase II,
dNTPs 0,25 mM, iniciadores IS5 e IS6 0,4 mM. 81 e
modelo de biblioteca de 15 µl, com o seguinte perfil térmico:
desnaturação inicial a 95 °C por 2 min, 14 ciclos de desnaturação a 95
°C por 30 s, recozimento a 60 °C por 30 s e alongamento a 72 °C por 30
s, seguido de um alongamento final a 72°C por 5 min. As bibliotecas
capturadas foram purificadas com colunas de rotação MinElute (QIAGEN) e
quantificadas com uma ScreenTape de alta sensibilidade D1000 em um
Agilent 2200 TapeStation.
Sequencing
For
both shotgun data retrieval and after the capture processing, the
samples were pooled in unimolar quantity (for SG sequencing up to 50
samples per pool, and for the capture process 2–8 samples per pool), and
sequenced on an Illumina NextSeq500 with 2 × 75 + 8 + 8 cycles
using the manufacturer’s protocols for multiplex sequencing at the
Functional Genomics Center in Zurich or at the Vienna BioCenter Core
Facilities.
Statistical analyses
Dataset selection
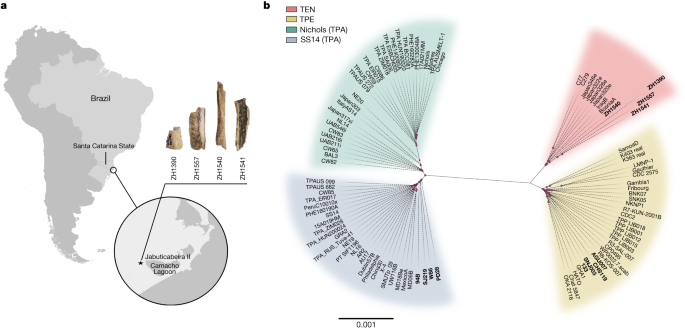
Montamos um conjunto de dados genômicos compreendendo 98 genomas de T. pallidum
disponíveis publicamente (8 TEN, 30 TPE e 60 TPA) de estudos publicados
anteriormente (incluindo 8 genomas antigos) e o recém-gerado genoma
ZH1540. Os genomas representam a variação genética das três
subespécies conhecidas de T. pallidum (TPA, TPE e TEN)
disponíveis até dezembro de 2022, e foram selecionados com foco em TEN e
TPE, devido à sua proximidade com o novo genoma antigo classificado
como TEN .
Os dados publicados para o conjunto de dados do genoma
moderno neste estudo estão disponíveis no banco de dados European
Nucleotide Archive (ENA): PRJNA313497 (números de acesso: SRR3268682 , SRR3268724 , SRR3268715 , SRR3268694 , SRR3268696 , SRR3268709 , SRR3268710 ), 11481 (números de acesso: ERR1470343 , ERR3596780 , ERR3596747 , ERR3596783 ), PRJEB28546 (números de acesso: ERR4045394 , ERR3684452 , ERR3684456 , ERR3684465 , SRR13721290 , ERR4853530 , ERR4993349 , ERR485358 7 , ERR4899206 , ERR5207017 , ERR5207018 , ERR5207019 , ERR4899215 , ERR4853623 , ERR4853625 ), PRJNA508872 (números de acesso: SRR8501165 , SRR8501164 , SRR8501167 , SRR8501166 , SRR8501168 , SRR8501171 ), PRJNA723099 (números de acesso: SRR14277267 , SRR14277266 , SRR14277458 , SRR14277444 ), PRJEB11481 (número de acesso: ERR 1470331 ), PRJDB9408 (números de acesso: DRR213712 , DRR213718 ), PRJNA588802 (números de acesso: SRR10430858 , SRS5636328 ), PRJNA322283 (número de acesso: SRR3584843 ), PRJNA754263 (números de acesso: SRR15440297 , SRR15440150 , SRR15440451 , SRR15440240 ), PRJEB40752 (números de acesso: ERR4690809 , ERR4690806 , ERR4690810 , ERR4690812 , ERR4690811
). Arquivos de montagem foram usados para 9 genomas do banco de
dados do National Center for Biotechnology Information (NCBI): CP002375.1 , CP002376.1 , NC_016842.1 , NC_017268.1 , NC_018722.1 , NC_021490.2 , NC_021508.1 , GCA_000813285.1 , CP035193.1 e para 24 genomas modernos do European Nucleotide Archive (ENA): CP021113.1 , CP073572.1 , CP073557.1 , CP073553.1 , CP073536.1 , CP073526.1 , CP073490.1 , CP073487.1 , CP07347 0 .1 , CP073447.1 , CP073446.1 , CP073399.1 , CP040555.1 , LT986433.1 , LT986434.1 , CP032303.1 , CP020366.1 , CP024088.1 , CP024089.1 , CP07812 , 1.1 , CP078090.1 , CP081507.1 , CP051889.1 e CP003902 .1 . Os dados de sequência bruta (arquivos fastq) usados para 6 genomas modernos estão disponíveis no banco de dados NCBI: PRJEB20795 (números de acesso: ERS1724928 , ERS1724930 , ERS1884567 ) e PRJNA343706 (números de acesso: SRR4308604 , SRR4308606 , SRR4308597 ). Genomas treponêmicos antigos publicados anteriormente aqui usados estão disponíveis na ENA: PRJEB37490 (número de acesso: ERR4065503 ), PRJEB37633 (número de acesso: ERR4000645 ), PRJEB35855 , PRJEB21276 (números de acesso: ERS2470995 , ERS2470994 ) e PRJEB62102 . Informações detalhadas sobre a fonte do conjunto de dados de referência estão documentadas na Tabela Suplementar 3 .
Selecionamos
todos os oito genomas TEN disponíveis publicamente, todos com mais de
99,4% de cobertura genômica, com exceção de C77 17 (81,4%). Selecionamos 30 genomas de TPE (Tabela Suplementar 3
). Para representar cada linhagem ou sub-linhagem, selecionamos pelo
menos um genoma, preferindo aqueles com maior profundidade de
sequenciamento e cobertura genômica. Todos os genomas TPE incluídos
têm mais de 95,3% de cobertura genômica, exceto os quatro antigos
genomas TPE: SJN003, AGU007, 133 e CHS119, exibindo 97,4%, 92,7%, 57% e
62% de cobertura genômica, respectivamente. Além disso, foram
incluídos 60 genomas de TPA das principais linhagens e sub-linhagens
descritas em estudos anteriores (Tabela Suplementar 3
). Todos esses genomas tinham mais de 90% de cobertura, exceto os
quatro genomas antigos, PD28, W86, SJ219 e 94B, todos com cobertura
genômica de 30% ou mais. Todos os genomas no conjunto de dados são
separados uns dos outros por pelo menos 5 SNPs. A cepa TPA Seattle-81
foi excluída do conjunto de dados final devido a mutações provavelmente
acumuladas durante passagens extensas em coelhos que podem causar
posicionamento ambíguo em filogenias 4 , 16 , 36 .
Os
dados brutos e/ou arquivos de montagem para cada genoma em nosso
conjunto de dados foram baixados dos bancos de dados públicos: European
Nucleotide Archive (ENA) 84 e Centro Nacional de Informações sobre Biotecnologia (NCBI) 85 . Os números de acesso são fornecidos na Tabela Suplementar 3 .
Processamento de leitura e geração de alinhamento de genoma baseado em múltiplas referências
To
reconstruct the individual genomes from the raw data, we carried out
raw read quality control and preprocessing, removing duplicates, variant
calling and filtering using the default parameters when not otherwise
specified. After processing the de-multiplexed sequencing reads, sample
sequencing quality was analysed with FastQC version 0.11.983, filtering reads with a QC value < 25. Following processing by cutadapt version 4.186
to remove the sequencing adapters, in order to reduce the reference
bias, and improve the posterior phylogenetic inference and assignment87,
the genome reference selection for mapping each sample was determined
according to the results from the original manuscript where the genomes
were published (see Supplementary Table 3). The mapping was carried out by BWA mem88(usando
parâmetros: -k 19, -r 2,5). Foram utilizados quatro genomas de
referência; os bem estudados genomas TEN e TPE BosniaA (NZ_CP007548.1)
e CDC2 ( NC_016848.1 ), bem como os genomas Nichols ( NC_021490.2 ) e SS14 ( NC_010741.1
), representando as duas linhagens principais dentro do TPA. No
entanto, para as novas amostras antigas obtidas aqui, os genomas de cada
amostra foram reconstruídos mapeando três genomas de referência de alta
qualidade, representando as três subespécies de T. pallidum (CDC2, BosniaA e Nichols).
CleanSam, do Picard Toolkit versão 2.18.29 ( http://broadinstitute.github.io/picard
), foi usado para limpar os arquivos SAM ou BAM fornecidos. As
leituras duplicadas foram removidas usando MarkDuplicates, do kit de
ferramentas Picard versão 2.18.29. AddOrReplaceReadGroups, do Picard
Toolkit versão 2.18.29, foi usado para atribuir todas as leituras em um
arquivo a um único novo grupo de leitura antes de usar mapDamage versão
2.2.0-86-g81d0aca 89
para estimar os parâmetros de dano ao DNA e redimensionar os índices de
qualidade de posições provavelmente danificadas nas leituras (usando o
parâmetro: --rescale).
Depois de gerar uma saída de pilha de texto para os arquivos BAM com a ferramenta mpileup do Samtools versão 1.7 90 , os SNPs foram chamados usando o VarScan versão 2.4.3 91 (usando
parâmetros: -p-valor 0,01, -min-reads2 1, -min-coverage 1,
-min-freq-for-hom, 0,4 -min-var-freq 0,05, -output-vcf 1). Em seguida,
uma filtragem SNP também foi realizada com VarScan (usando para as
amostras modernas os parâmetros: -p-value 0,01, -min-reads2, 5
-min-coverage 10, -min-avg-qual 30 -min-freq-for -hom 0,4, -min-var-freq
0,9, -output-vcf 1 e modificação de alguns parâmetros para as amostras
antigas devido à sua menor cobertura e qualidade de leitura: -p-value
0,01 -min-reads2 3, -min-coverage; 5, -min-avg-qual 30,
-min-freq-for-hom 0,4, -min-var-freq 0,9 -output-vcf 1). Além disso,
todas as posições com menos de 3 leituras mapeadas foram mascaradas com
Genomecov do Bedtools versão 2.26.0 92 para
amostras modernas e antigas. Todas as etapas de geração do genoma
foram visualizadas e confirmadas manualmente com Tablet versão
1.21.02.08 93 ,
verificando cada SNP um por um e descartando os possíveis SNPs espúrios
do novo genoma antigo ZH1540. As sequências finais resultantes foram
obtidas por maskfasta do Bedtools v2.26.0.
Além disso, utilizamos metodologias testadas de sequenciamento e análise posterior 17 , 42 para obter maior cobertura e genomas modernos de T. pallidum mais confiáveis . Sempre que possível, foram obtidos arquivos de montagem em vez de dados brutos (Tabela Suplementar 3 ). Um alinhamento de genoma baseado em referência múltipla para todas as sequências foi gerado no MAFFT v7.467 94 (usando
parâmetros: --adjustdirection --auto --fastaout --reorder). Porém,
devido ao uso de diferentes referências genômicas, regiões com baixa
cobertura para alguns genomas, correspondendo principalmente aos genes tpr e arp , foram revisadas e alinhadas manualmente com o Aliview versão 1.25. 95 .
As
amostras ZH1390, ZH1541 e ZH1557 tinham dados suficientes para tentar
uma reconstrução do genoma e foram determinadas como tendo o maior
número de SNPs em comum com a referência TEN, mas foram excluídas das
análises downstream devido à cobertura limitada adquirida para cada uma
delas, o que fez os SNPs obtidos são menos confiáveis. A amostra
ZH1540, no entanto, rendeu uma cobertura genômica notável de 33,6× e foi
selecionada para análises aprofundadas subsequentes.
Proteinorto versão 6.0b 96
(usando parâmetros: -p=blastn -singles -keep) foi usado para conduzir
um estudo de ortologia a fim de encontrar genes ortólogos nos quatro
genomas de referência utilizados 96 .
Cada gene presente em pelo menos um dos quatro genomas de referência
teve suas coordenadas genômicas determinadas com base em sua localização
no alinhamento final mesclado (ver Tabela Suplementar 3 ).
Para
verificar a precisão do alinhamento final do genoma múltiplo, e que
nenhum gene codificador de proteína foi inadvertidamente truncado, as
traduções de proteínas para cada gene presente em pelo menos um genoma
de referência foram comparadas com os arquivos gff3 originais de cada
uma das quatro referências (Suplemento Tabela 3
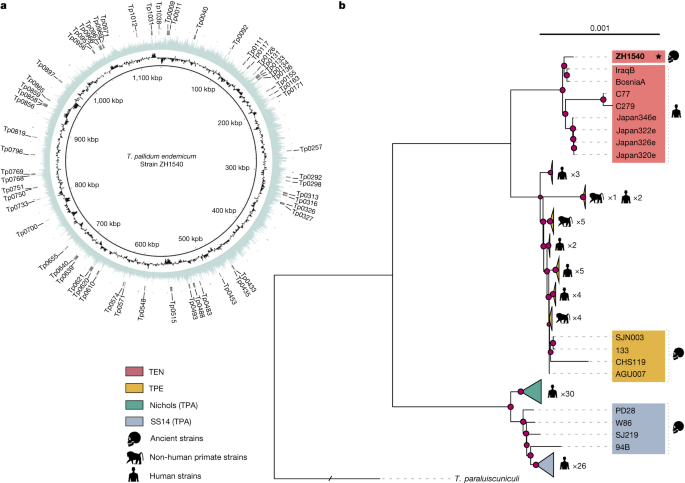
). O genoma ZH1540 reconstruído e suas principais características
foram representados graficamente usando BRIG versão 0.95-dev.0003 97 .
Análise de recombinação usando PIM
Como observado anteriormente 36 , a presença de recombinação nos genomas de T. pallidum
pode interferir nas topologias das árvores filogenéticas inferidas.
Para analisar a potencial recombinação genética, usamos o pipeline PIM 36 para detectar recombinação gene por gene. Em resumo, o processo envolveu as seguintes etapas:
-
(1)
Using IQ-TREE version 1.6.10, a maximum-likelihood tree was created for the multiple genome alignment98. All maximum-likelihood trees for the remaining steps were obtained using GTR99 + G100 + I101 as an evolutionary model and 1,000 bootstraps replications.
-
(2)
The 1,161 genes found in at least one of the
reference genomes were extracted, and the number of SNPs for each gene
was calculated. Genes with less than three SNPs were excluded.
-
(3)
The phylogenetic signal in each gene alignment for each of the remaining genes was evaluated by likelihood mapping102 in IQ-TREE (using parameters: -lmap 10000 -n 0), retaining only those genes that showed a phylogenetic signal.
-
(4)
A maximum-likelihood tree was generated for each of the remaining genes using IQ-TREE.
-
(5)
Para cada gene incluído, testamos a
congruência filogenética entre árvores usando IQ-TREE (usando
parâmetros: -m GTR + G8 -zb 10000 -zw), comparando a árvore de máxima
verossimilhança obtida a partir do alinhamento do gene e a árvore de
máxima verossimilhança obtida do alinhamento do genoma completo usando
dois métodos diferentes: Shimodaira – Hasegawa 103 e pesos de verossimilhança esperados (ELW) 104 .
Genes para os quais pelo menos um teste rejeitou a topologia da
árvore de referência com o alinhamento do gene adotando uma abordagem
conservadora ( P <0,2, valor de peso próximo a 0, para os
testes Shimodaira-Hasegawa e ELW, respectivamente) e o alinhamento
completo do genoma rejeitou a topologia de as árvores construídas
utilizando o alinhamento gênico (incongruência recíproca, P < 0,2 e valor de peso próximo a 0) em pelo menos um deles foram selecionadas e examinadas mais de perto na etapa seguinte.
-
(6)
Usando MEGAX 105 ,
os genes selecionados que apresentavam incongruência recíproca foram
posteriormente examinados para avaliar e descrever potenciais eventos de
recombinação. Um gene deve ter pelo menos três SNPs homoplásicos
próximos – SNPs que são compartilhados por vários grupos (TPE, TEN,
TPA-Nichols ou TPA-SS14) e produzir uma distribuição polifilética – para
ser rotulado como recombinante. Os SNPs homoplásicos encontrados no
alinhamento dos genes serviram como limites das áreas recombinantes.
-
(7)
Utilizando um critério de parcimônia na
distribuição de estados alternativos dos SNPs homoplásticos, foram
inferidos os potenciais clados ou cepas doadoras e receptoras de cada
evento de recombinação.
DNA sections, a number of genes have a high
percentage of sites with missing data. The majority of these genes are
members of the tpr and arp families, which include
collections of paralogous genes. In order to continue analysing these
intriguing genes with the PIM pipeline, strains that had a high
percentage of missing data in each of these genes were eliminated.
Following previous findings35,36, the hypervariable gene tprK (tp0897), with seven hypervariable regions that undergo intrastrain gene conversion17,37,106,107,108,109, and the tp0316 and tp0317 genes, also under gene conversion, were completely excluded from the recombination analysis.
PIM procedure for likelihood mapping and topology tests
Um teste de mapeamento de verossimilhança foi realizado usando IQ-TREE para determinar quais genes (Tabela Suplementar 4
) mostraram um sinal filogenético (dos 382 genes para os quais> 3
SNPs foram encontrados em comparação pareada com pelo menos um genoma de
referência). Para cada quarteto (subconjunto de quatro sequências)
nos dados, o teste cria árvores filogenéticas não enraizadas. As
probabilidades do quarteto são então traçadas dentro de um triângulo,
onde a posição denota a “semelhança em árvore” do quarteto em questão.
Os quartetos de canto estão completamente resolvidos, os quartetos nas
laterais estão parcialmente resolvidos e os quartetos no centro não
estão resolvidos. Dos 382 genes, 29 tinham muitos valores faltantes
para serem testados usando o método de mapeamento de verossimilhança.
Para incluir esses genes nas próximas etapas do pipeline PIM e nas
comparações de topologia, as sequências problemáticas com mais de 50% de
posições com dados faltantes foram removidas.
Após o teste de
mapeamento de verossimilhança, 9 genes pertencentes à zona central do
triângulo foram descartados (Tabela Suplementar 4
). Em seguida, usando os testes de topologia Shimodaira-Hasegawa e
ELW, comparamos as árvores gênicas dos genes restantes com a árvore de
referência preliminar do alinhamento do genoma completo para avaliar sua
congruência filogenética (Tabela Suplementar 4
). Dos 373 genes que testaram positivo para incongruência
filogenética, 27 continham pelo menos três SNPs consecutivos, apoiando
um evento de recombinação. A estes adicionamos tp0859 , que foi detectado como recombinante em um estudo anterior 35 , resultando em um total de 27 genes recombinantes.
Análise de recombinação usando Gubbins e ClonalFrameML
Gubbins version 2.3.1110 and ClonalFrameML version 1.11-1111
are frequently used tools for the genome-wide identification of
recombinant positions in bacterial genomes. To test the robustness of
our recombination analysis using PIM, we also ran these two programs,
with default parameters and the same whole-genome alignment used with
PIM. Gubbins identified 301 distinct recombination events associated
with 103 genes, ranging in size from 5 bp to 13,866 bp. Similarly,
ClonalFrameML detected 656 events, with 32 of them being 1 or 2 bp long,
and the longest event spanning 782 bp. Notably, all the genes
identified by PIM as having a recombinant region were also detected by
both ClonalFrameML and Gubbins, except for gene tp0558, which was missed by ClonalFrameML but detected by Gubbins. Additionally, genes tp0164 and tp0179 were detected by ClonalFrameML but missed by Gubbins.
Phylogenetic analysis
Uma
árvore de máxima verossimilhança baseada no alinhamento incluindo todos
os genes foi construída com IQ-TREE, usando GTR + G + I como modelo
evolutivo e 1.000 replicações de bootstrap (Extended Data Fig. 2a
). Em seguida, os genes identificados como recombinantes pelo PIM
foram removidos do alinhamento do genoma múltiplo. Três genes
adicionais ( tp0897 , tp0316 e tp0317 ), que
contêm regiões repetitivas e foram identificados como hipervariáveis
e/ou sob conversão gênica no passado, também foram removidos para
evitar a introdução de um viés potencial. Como o gene tp0317 está aninhado dentro do gene tp0316 e as coordenadas do genoma de referência da BósniaA para tp0316 cobriram uma área maior do que as dos outros genomas de referência, tp0316 e tp0317 foram removidos de acordo com as coordenadas tp0316
do genoma de referência da BósniaA. Uma árvore filogenética de
referência foi então construída empregando o novo alinhamento do genoma
de herança vertical, também com IQ-TREE usando GTR + G + I como modelo
evolutivo e 1.000 replicações de bootstrap (Extended Data Fig. 2b ). Ambas as árvores obtidas foram comparadas e são mostradas na Fig. 2 de dados estendidos .
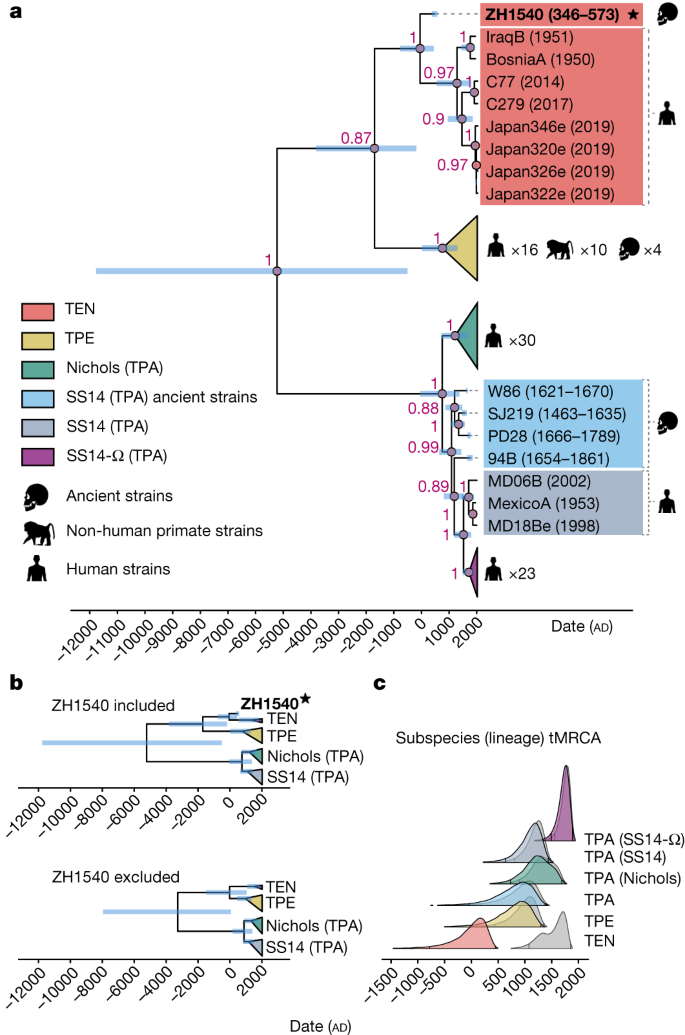
A
linhagem SS14 foi descrita anteriormente como um grupo amplamente
epidêmico e resistente a macrolídeos que surgiu após, e possivelmente
foi motivado pelo, uso clínico de antibióticos após sua descoberta. 12 , 16 .
Com base nos resultados da nossa análise filogenética e expandindo as
classificações filogenéticas anteriores e a nomenclatura da linhagem
SS14 12 , 16 ,
definimos o clado que contém quase todos os genomas SS14 de amostras
clínicas e contemporâneas como a sub-linhagem SS14-Ω. No entanto, duas
amostras clínicas contemporâneas (MD18Be e MD06B) não foram
classificadas como sub-linhagem SS14-Ω, porque essas amostras se agrupam
com o genoma do MéxicoA, em linha com resultados publicados
anteriormente 42.
To
compare the PIM-based analysis with other widely used recombination
detection methods, Gubbins and ClonalFrameML, we followed a similar
procedure of removing the recombinant positions detected by these tools
and inferred maximum-likelihood trees with the retained positions in the
corresponding multiple genome alignments. All the phylogenetic trees
with recombination events removed exhibit general congruence with each
other, whether the events were identified by PIM, Gubbins or
ClonalFrameML. Furthermore, the placement of the ZH1540 genome remained
consistent in the phylogenetic trees, regardless of the recombination
detection method employed, and despite the elimination of recombinant
genes to generate the vertically inherited alignment.
Exploratory characterization of the 16S-23S genes
T. pallidum contains two rRNA (rrn)
operons, each of which encodes the 16S-23S-5S rRNA genes and intergenic
spacer regions (ISRs). There is evidence that the random distribution
of rrn spacer patterns in T. pallidum may be generated by reciprocal translocation of rrn operons mediated by a recBCD-like system found in the intergenic spacer regions (ISRs)112 . De acordo com estudos anteriores 112 , 113 , 114 , 115 , descobrimos que os ISRs 16S-23S das cepas de TPA contêm os genes tRNA-Ile (tRNA-Ile-1; tp0012 ) e tRNA-Ala (tRNA-Ala-3; tp00t15 ) dentro dos rrn1 e rrn2 operons , respectivamente . Por outro lado, os genomas TPE mostram um padrão espaçador Ala/Ile, onde os tp0012 e tp00t15 ortólogos estão localizados dentro dos operons rrn2 e rrn1 , respectivamente.
Identificamos 68 SNPs nos genes r0001 , r0002 , r0004 e r0005
, codificando os genes 16S-23S rRNA do novo e antigo genoma ZH1540,
colocando-os entre os genes mais variáveis em nosso alinhamento e
aumentando o potencial de que incluí-los no alinhamento poderia resultar
em uma reconstrução filogenética tendenciosa. Embora os SNPs
encontrados pareçam ser bem suportados pelas leituras obtidas a partir
do mapeamento de sequências (Tabela Suplementar 3
), a sua origem de possível contaminação não pode ser completamente
descartada e seriam necessárias análises adicionais para os confirmar.
A exclusão destes genes do alinhamento, além dos genes recombinantes e tp0316, tp0317 e tp0897 , não resultou em quaisquer alterações na topologia (Extended Data Figs. 2 b e 3
), embora os comprimentos dos ramos tenham sido alterados. Como se
sabe que esses genes possuem regiões conservadas, além de regiões
variáveis, usadas para explorar as relações evolutivas entre bactérias
patogênicas 116 , 117 , 118 ,
decidimos mantê-los no alinhamento para todas as análises subsequentes.
Finalmente, notamos que o genoma ZH1540 não possuía nenhuma das duas mutações do gene de RNA ribossômico 23 S de T. pallidum
conhecidas por conferir resistência a macrólidos (A2058G e A2069G).
Em contraste, quatro estirpes modernas de TEN do Japão possuem a mutação
A2048G, sugerindo uma recente pressão de selecção para mutações de
resistência a antibióticos.
Molecular clock dating
We used the Bayesian phylogenetics package BEAST2 v2.6.7119 to estimate a time-calibrated phylogeny of the context dataset of 98 T. pallidum
genomes along with our new ancient genome, ZH1540. We removed
hypervariable and recombining genes from the alignment, as described
above, reduced it to variable sites and used an ascertainment bias
correction to account for constant sites.
Análises de regressão da raiz às pontas (Dados Estendidos Fig. 4
) mostram que, embora haja uma correlação positiva entre o ano de
amostragem e a divergência da raiz às pontas entre todas as cepas
clínicas modernas, indicando uma evolução semelhante a um relógio, a
correlação é muito fraca quando incluindo também cepas passadas e
negativo quando incluindo cepas antigas. Dentro dos clados TPE, TEN e
SS14 existe uma correlação positiva entre todas as cepas clínicas
modernas e passadas. Por outro lado, a correlação é negativa para
cepas de Nichols, mesmo quando se olha apenas para cepas clínicas. A
fim de levar em conta a variação da taxa e os longos ramos terminais em
algumas cepas (provavelmente devido a uma infinidade de efeitos,
incluindo erros de sequenciamento, contaminação e mutações introduzidas
durante a passagem do coelho), usamos um modelo de relógio UCLD e UCED
para a datação do relógio molecular. análise 120 . Para ambos os modelos, colocamos um prior lognormal estreito com uma média (no espaço real) de 1 × 10 −7 substituições
por local por ano e desvio padrão de 0,25 na frequência média do clock.
Este forte anterior foi usado para compensar o fraco sinal temporal
entre os genomas de T. pallidum e foi calibrado em estimativas anteriores da taxa de substituição 4 , 35 . Usamos ainda um modelo de substituição GTR + G + I 118 e um gráfico do horizonte bayesiano 121 modelo
demográfico (árvore anterior) com 10 grupos. Para todos os genomas
onde as datas de amostragem não são conhecidas exatamente, usamos
anteriores uniformes ao longo dos intervalos de datas relatados nos
estudos originais para levar em conta a incerteza 4 , 5 , 6 , 16 , 122 . Para ZH1540, definimos o intervalo de datas para 364–573 dC
, de acordo com os resultados da datação por radiocarbono corrigidos
pelo efeito do reservatório marinho acima. Priores padrão foram usados
para todos os outros parâmetros do modelo. A mesma análise foi
repetida sem ZH1540, a fim de avaliar o efeito do nosso novo genoma
antigo nas datas de divergência. Repetimos ainda mais a análise usando
um lognormal anterior amplo com uma média (no espaço real) de 1 × 10 −7
substitutions per site per year and standard deviation 1 on the mean
clock rate and using both constant-size and exponential growth
coalescent models to assess the impacts of the mean clock rate prior and
demographic models on divergence time estimates.
Para cada análise, executamos quatro cadeias de Markov Monte Carlo (MCMC) de 5 × 10 8 steps each, sampling parameters and trees every 10,000 steps. After assessing convergence in Tracer v1.7123
and confirming that all four chains converged to the same posterior
distribution, we combined the chains after discarding the first 10% of
samples as burn-in. In the resulting combined chains all parameters have
effective sample size (ESS) values > 150. TreeAnnotator v2.6.7 was
used to compute MCC trees and the results were visualized using ggplot2124, ggtree125
and custom scripts. The 95% HPD of the coefficient of variation
estimated under the UCLD model excluded 0 (median = 1.46, 95% HPD
1.08–1.9), indicating that a strict clock model is not appropriate for
our dataset. Robustness analyses show that under a narrow mean clock
rate prior both the UCED and UCLD clock models result in similar
divergence time estimates (Extended Data Fig. 5a–f),
with the UCED model estimates tending to be more recent and the UCLD
model estimates usually having longer tails. Under a wide mean clock
rate prior, estimates with the UCED are broadly similar, albeit wider,
while the UCLD model estimates very wide posterior distributions for
divergence times, indicating little information under this model.
Divergence time estimates were not sensitive to the demographic model
used. The MCC trees under the UCED model with a narrow prior, both with
and without ZH1540 included in the analysis are shown in Extended Data
Figs. 6 and 7, respectively.
Finalmente, realizamos um teste bayesiano de randomização de datas 126 , 127 , 128 (DRT)
para avaliar melhor a força do sinal temporal em nosso conjunto de
dados, permutando datas de amostragem entre genomas e realizando 50
análises replicadas. Para as análises, utilizou-se o conjunto de dados
completo, um modelo de relógio UCED com um anterior estreito e o modelo
demográfico Bayesiano do horizonte, fixando as datas de amostragem de
cepas antigas às médias dos intervalos de datas de radiocarbono para
simplificar. Cadeias MCMC foram executadas por 1 × 10 8 passos, amostragem de parâmetros a cada 10.000 passos. A convergência foi avaliada usando a coda 129 pacote
para garantir que todos os parâmetros em todas as cadeias tenham
valores de ESS > 150. Os resultados do DRT mostram que os intervalos
HPD de 95% da taxa de clock média em réplicas com datas de amostragem
permutadas são muito menores do que o esperado se todas as informações
vierem da taxa de clock média anterior (Dados Estendidos Fig. 5g
). Em geral, os intervalos HPD não se sobrepõem ao intervalo HPD de
95% da taxa média de clock estimada com as datas de amostragem
verdadeiras.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.