Redefinindo a história treponêmica através de genomas pré-colombianos do Brasil
Nature volume 627 , páginas 182–188 ( 2024 )
Abstrato
As origens das doenças treponémicas permanecem desconhecidas há muito tempo, especialmente considerando o início repentino da primeira epidemia de sífilis no final do século XV na Europa e a sua hipotética chegada das Américas com as expedições de Colombo. 1 , 2 . Recently, ancient DNA evidence has revealed various treponemal infections circulating in early modern Europe and colonial-era Mexico3,4,5,6. No entanto, até onde sabemos, não há nenhuma evidência genómica de treponematose recuperada nas Américas ou no Velho Mundo que possa ser datada de forma fiável como antes dos primeiros contactos transatlânticos. Aqui, apresentamos genomas treponêmicos de restos humanos de quase 2.000 anos de idade no Brasil. Nós reconstruímos quatro genomas antigos de um patógeno treponêmico pré-histórico, mais intimamente relacionado ao agente causador do bejel, Treponema pallidum endemicum . Contradizendo o nicho geográfico moderno do bejel nas regiões áridas do mundo, os resultados questionam a caracterização paleopatológica anterior das subespécies de treponemas e mostram o seu potencial adaptativo. Um genoma de alta cobertura é usado para melhorar as estimativas de data do relógio molecular, colocando firmemente a divergência das subespécies modernas de T. pallidum em tempos pré-colombianos. No geral, nosso estudo demonstra as oportunidades dentro da arqueogenética para descobrir eventos-chave na evolução e emergência de patógenos, abrindo caminho para novas hipóteses sobre a origem e disseminação das treponematoses.
Conteúdo semelhante sendo visualizado por outras pessoas

As infecções treponêmicas, causadas pela bactéria T. pallidum , estão aumentando a taxas alarmantes em todo o mundo 7,8,9,10,11. Increasing evidence suggests that many treponemal strains have developed antibiotic resistance, which is expected to facilitate their spread12. This re-emerging threat has led to many modern genetic and medical studies8,13,14,15. intimamente relacionadas de T. pallidum As subespécies T. pallidum pallidum (TPA), T. pallidum pertenue (TPE) e T. pallidum endemicum (TEN) - responsáveis pela sífilis, bouba e bejel, respectivamente - têm sequências genômicas altamente semelhantes que diferem em aproximadamente 0,03% 16 , 17 .
Hoje, o bejel está geograficamente concentrado em ambientes áridos e quentes, especialmente no Mediterrâneo oriental e no oeste da Ásia, enquanto a bouba é encontrada principalmente nos trópicos úmidos e quentes, como a África ou a América do Sul. 18 . Entre as treponematoses, a sífilis é a mais distribuída globalmente; é comum mesmo em populações ocidentais ricas com fácil acesso a cuidados de saúde 7 , 13 . Por outro lado, a bouba e o bejel afetam principalmente os países em desenvolvimento e permanecem menos extensivamente estudados 18 .
Historicamente, a sífilis venérea é conhecida por ter causado um surto devastador na Europa no final do século XV. Os sintomas que podem desenvolver-se na ausência de tratamento eficaz incluem desfiguração física grave, cegueira e deficiência mental. 19 . Como manifestações semelhantes podem aparecer em todas as treponematoses 20 , 21 , 22 , a sua distinção ao nível da subespécie é muitas vezes pouco fiável e baseia-se principalmente na localização de úlceras cutâneas características (nos órgãos genitais ou noutros locais), especialmente em países em desenvolvimento com recursos médicos limitados 23 , 24 , 25 . Os diagnósticos de casos históricos são igualmente difíceis: embora as treponematoses possam deixar alterações patológicas nos ossos, estas aparecem em apenas aproximadamente 5–30% dos casos avançados. 26 , 27 , resultando em provável subestimação da prevalência passada de treponematoses.
The early presence and potential origin of syphilis in Europe was proposed in the pre-Columbian hypothesis, based on osteological analyses of treponemal lesions, whereas the Columbian hypothesis associates its emergence with Columbus’ first American expedition and considers the contradicting palaeopathological evidence to be unreliable2. Before the distinctions among the subspecies could be genetically defined28,29, the unitarian hypothesis claimed that all treponematoses were the same disease, which only manifested differently under different environmental and cultural factors. Attempts to identify subspecies using palaeopathology have had ambiguous results and require DNA evidence as confirmation: previous ancient DNA studies have, for example, revealed that some cases of presumed syphilis instead correspond to yaws4,5, and recovered at least one previously unknown T. pallidum strain4. Since treponemes possess an impressive ability to adjust to various environments and are known to have previously occupied geographical regions outside their present distributions4,5, apenas evidências inequívocas de DNA treponêmico pré-colombiano podem iluminar as origens da sífilis e também desvendar aspectos importantes da história evolutiva de todos os treponemas.
Aqui, apresentamos evidências de doença treponêmica pré-colombiana no Novo Mundo a partir de um cemitério de sambaqui brasileiro com quase 2.000 anos de idade, Jabuticabeira II, através de quatro genomas reconstruídos de T. pallidum com cobertura de até 33,6x, filogeneticamente basal ao diversidade moderna da subespécie causadora de bejel, T. pallidum endemicum .
Origens geográficas e paleopatologia
Noventa e nove exemplares de Jabuticabeira II da região de Laguna de Santa Catarina, na costa brasileira, com e sem patologias, foram incorporados neste estudo. Análises osteológicas anteriores revelaram patologias relacionadas com infecções, sugerindo potenciais infecções treponémicas 30 , como periostite (24 casos), remodelação óssea (4 casos) e marcas de traça no crânio (4 casos). Das 37 amostras consideradas preliminarmente positivas para DNA treponêmico após a triagem inicial, 12 eram de indivíduos com patologias e o restante era proveniente de espécimes não patológicos (Tabela Suplementar 1 ). Quatro amostras ósseas, de quatro indivíduos diferentes, produziram dados genômicos suficientes para análises abrangentes. A amostra ZH1390 (Tabela 1 e Fig. 1a ) representa um fragmento de tíbia apresentando periostite. A amostra ZH1540 veio de um conjunto de ossos misturados de um esqueleto incompleto, nomeadamente de uma fíbula com lesões patológicas (Tabela 1 e Fig. 1a ). As amostras ZH1541 e ZH1557 originaram-se de ossos longos sem nenhuma patologia identificada (Tabela 1 e Figura 1a ). Todas as amostras foram datadas por radiocarbono e testadas quanto ao efeito de reservatório marinho. Os dados brutos, calibrados e corrigidos de 14 A datação C é apresentada em Dados Estendidos Fig. 1a, b , Tabela Suplementar 2 e Métodos , 'Informações Arqueológicas'. Os indivíduos positivos para DNA treponêmico não foram enterrados separadamente dos demais indivíduos em Jabuticabeira II, sugerindo que foram tratados igualmente.
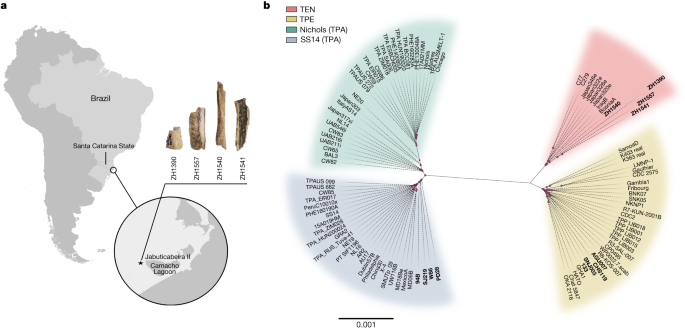
a , Mapa mostrando a localização do sítio arqueológico Jabuticabeira II, no litoral sul do estado de Santa Catarina, Brasil, e as amostras ZH1390, ZH1540, ZH1541 e ZH1557, cujos genomas foram reconstruídos. b , Uma árvore filogenética de máxima verossimilhança das cepas modernas e antigas de T. pallidum usando GTR + G + I (ver Métodos ) como modelo evolutivo e 1.000 repetições de bootstrap. Todos os genomas antigos utilizados neste estudo (genomas antigos recentemente reconstruídos e publicados anteriormente; ver Tabela Suplementar 3 ) estão marcados em negrito. Os pontos rosa representam nós com valores de bootstrap superiores a 70%. A barra de escala está em unidades por substituições por site.
Preliminary pathogen screening
Na triagem inicial a partir de dados de sequenciamento shotgun, 37 de 99 amostras mostraram entre 7 e 133 ocorrências para táxons da família Treponema no banco de dados Kraken e foram incluídas no processo de enriquecimento de alvo (Tabela Suplementar 1 e Métodos , 'Processamento de amostra') . Destas amostras, 9 tiveram mais de 5.000 leituras mapeadas para 3 genomas de referência de T. pallidum (BosniaA, CDC2 e Nichols) pós-captura e foram, portanto, consideradas positivas para infecção treponêmica (Tabela Suplementar 1 ). Para estas amostras positivas, três bibliotecas adicionais de cadeia dupla foram produzidas para uma segunda rodada de enriquecimento de todo o genoma 4 , 31 (para uma metodologia detalhada, consulte Métodos , 'Processamento de amostras'). Após o enriquecimento adicional, os dados de todas as bibliotecas feitas a partir dos mesmos extratos originais e todos os dados de sequenciamento produzidos nas duas rodadas de captura foram combinados para cada amostra. Quatro amostras, ZH1390, ZH1540, ZH1541 e ZH1557, tiveram leituras cobrindo 9,2–99,4% do genoma de referência da BósniaA em 1×, com uma cobertura média entre 2× e 33,6× (Tabela 1 ). Essas quatro amostras foram consideradas como tendo maior potencial para reconstrução do genoma completo e análises posteriores.
Estimativa de autenticidade de DNA antigo
A autenticidade do DNA antigo foi confirmada examinando a desaminação de bases nas extremidades das leituras: 21%, 10%, 12% e 7% nas extremidades 5' e 17%, 12%, 14% e 6% nas extremidades 3. ′ termina para as amostras ZH1390, ZH1540, ZH1541 e ZH1557, respectivamente (Extended Data Fig. 1c – f ). As amostras tinham comprimentos médios de fragmentos 32 , 33 , 34 variando de 64 pb a 74 pb (Tabela 1 ). Além disso, a atribuição dos cromossomos sexuais dos dados do shotgun foi consistente com XX para as amostras ZH1540 e ZH1541. Embora os indivíduos tenham sido anteriormente considerados prováveis do sexo masculino nas análises osteológicas, as amostras ZH1390 e ZH1557 produziram dados insuficientes para a determinação molecular do sexo (Tabela 1 e Métodos , 'Informações arqueológicas').
Reconstrução do genoma
After high-throughput Illumina sequencing of the enriched DNA from the 4 selected samples, the resulting 20–100 million raw reads were merged sample-wise and duplicate reads were removed (Table 1). Genomes were reconstructed by mapping each sample to three representative high-quality reference genomes of T. pallidum subspecies: CDC2 for TPE, BosniaA for TEN, and Nichols for TPA (Methods, ‘Sample processing’ and ‘Dataset selection’). We filtered positions on the basis of read coverage, variant allele frequency, P value and base quality, and obtained three different consensus sequences for each sample, each with a different number of covered bases, as well as SNPs. The number of SNPs in each sequence, along with the phylogenetic analyses consistently supported a placement of all four samples within the TEN clade (Figs. 1b and 2b, Table 1 and Supplementary Table 3). Although the consensus sequences from three samples, ZH1390, ZH1541 and ZH1557, were assigned to T. pallidum endemicum (Fig. 1b), read coverage was below the threshold required for downstream analyses (for details, see Methods ‘Sample processing’ and ‘Read processing and multiple reference-based genome alignment generation’).
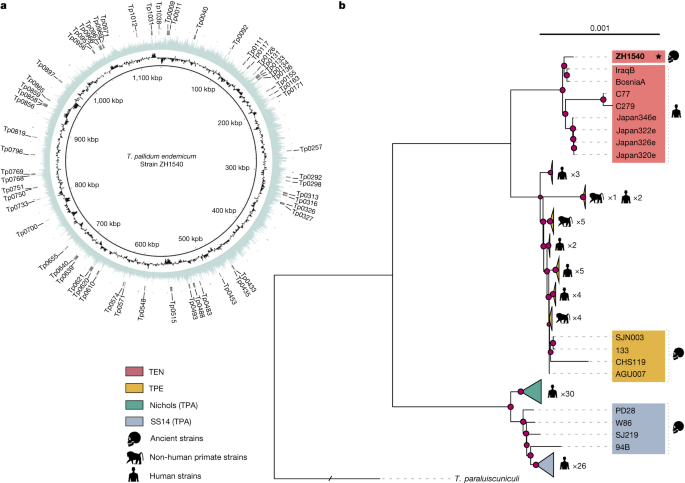
a , Gráfico circular do genoma ZH1540. Os círculos indicam (de dentro para fora): posição genômica, conteúdo de GC (preto) e cobertura (azul). A borda externa (cinza) mostra um conjunto de 60 genes candidatos associados à virulência e descritos em estudos anteriores 3,4. b , Árvore filogenética de máxima verossimilhança recolhida obtida usando o alinhamento do genoma completo e usando Treponema paraluiscuniculi para enraizar a árvore usando GTR + G + I (ver Métodos ) como modelo evolutivo e 1.000 repetições de bootstrap. O novo genoma antigo ZH1540 está destacado em negrito e com uma estrela. As sub-linhagens dentro de cada subespécie ou linhagem são recolhidas, com exceção dos genomas antigos e do clado TEN. Os pontos rosa representam nós com valores de bootstrap superiores a 70%.
A sequência final obtida para a amostra ZH1540 resultou em 99,38% de cobertura em relação ao genoma de referência TEN (BósniaA), uma profundidade de cobertura mínima de 5× e uma profundidade mediana de 33,6× (Tabela 1 e Fig. 2a ). A chamada de variante resultou na identificação de 123 SNPs, cada um dos quais foi verificado individualmente (detalhes fornecidos na Tabela Suplementar 3 e em Métodos , 'Processamento de amostra' e 'Processamento de leitura e geração de alinhamento de genoma baseado em múltiplas referências'). Das referências modernas disponíveis, o novo genoma antigo da TEN exibe uma diferença de 123 SNPs em comparação com as amostras da BósniaA e do IraqueB. No entanto, o número de posições diferentes é muito maior em comparação com os 4 genomas RTE japoneses (205 SNPs) e os genomas RTE cubanos (504 SNPs).
Alinhamento do genoma baseado em múltiplas referências
The new ancient genome ZH1540 was analysed together with an additional 98 publicly available genomes, including 8 modern TEN strains, 30 TPE strains (including 9 genomes from primates and 4 ancient genomes), 30 Nichols-lineage and 30 SS14-lineage TPA strains (including 4 ancient genomes) (Supplementary Table 3). Assembly files for 33 of these 98 genomes were available and downloaded directly from the public databases European Nucleotide Archive (ENA) and National Center for Biotechnology Information (NCBI). For the remaining 65 genomes, we mapped the raw sequencing data to the closest of four representative reference genomes (CDC2, BosniaA, Nichols and SS14), to obtain new assembly files. The genome reference selected for each sample was based on the subspecies and/or lineage classification of each sample from the original publications (Supplementary Table 3). A multiple reference-based genome alignment of 98 sequences from several sources was generated according to the previously published methodology35. O alinhamento resultante abrangeu um total de 1.141.812 nucleotídeos com 6.149 SNPs detectados (consulte Disponibilidade de dados e métodos , 'Processamento de amostras' e 'Processamento de leitura e geração de alinhamento de genoma baseado em referências múltiplas').
Phylogenetic and recombination analyses
Uma reconstrução filogenética confiável exigiu a remoção de regiões genômicas herdadas não verticalmente, como regiões recombinantes ou loci com conversão intra ou intergênica. Em uma análise de recombinação com o método de incongruência filogenética 36 (PIM), we detected 34 recombinant regions across 27 genes, encompassing a total of 957 SNPs (15.56% of the total SNPs) (Supplementary Table 3 and Supplementary Table 4). Owing to the exclusion of the highly passaged Seattle-81 strain, 3 of the previously detected recombinant genes were not detected here, and 11 detected genes were novel in relation to the previously published results. The average length of the recombinant regions was 368 bp, with a minimum length of 4 bp and a maximum of 2,209 bp. Notably, all the recombination events detected here correspond to inter-subspecies transfers with the exception of an intra-subspecies recombination event found in the tp0117 gene and three additional genes for which the putative donors are unidentified external sources (Supplementary Table 4 and Methods, ‘Recombination analysis using PIM’ and ‘PIM procedure for likelihood mapping and topology tests’).
Para construir um alinhamento de herança estritamente vertical, removemos os 27 genes recombinantes detectados aqui junto com três genes, tp0316 , tp0317 e tp0897 , que são conhecidos por serem hipervariáveis e/ou sujeitos à conversão gênica 37 , 38 , desde o alinhamento inicial (ver Disponibilidade de dados e métodos , 'Análise filogenética'). O alinhamento final livre de recombinação abrangeu 1.103.436 pb com 3.718 SNPs. Árvores de máxima verossimilhança foram construídas usando alinhamentos múltiplos de genoma (Fig. 2b e Dados Estendidos Figs. 2 e 3 ). Na Fig. 2 de dados estendidos , as topologias das duas árvores de máxima verossimilhança com e sem os loci recombinantes ou hipervariáveis são comparadas.
A eliminação de genes herdados não verticalmente teve um efeito menor na reconstrução da filogenia de T. pallidum (Extended Data Figs. 2b e 3 e Methods , 'Phylogenetic Analysis'). Os resultados após a remoção dos sítios recombinantes detectados no PIM foram confirmados com dois outros programas de detecção de recombinação, Gubbins e ClonalFrameML ( Methods , 'Recombination analysis using Gubbins and ClonalFrameML'). Além disso, foram avaliadas mutações genéticas (A2058G e A2059G) relacionadas à resistência a antibióticos macrólidos 12 , e foram encontrados ausentes no genoma antigo ZH1540 (Methods, 'Caracterização exploratória dos genes 16S-23S').
Molecular clock dating
A datação do relógio molecular foi realizada no mesmo conjunto de dados acima, com 27 genes recombinantes, tp0316 , tp0317 e tp0897 removidos. Na filogenia calibrada no tempo estimada, todas as três subespécies (TEN, TPE e TPA), bem como as linhagens SS14 e Nichols de TPA receberam alto suporte para formar clados distintos (probabilidade posterior> 0,97; Fig. 3a e Tabela de Dados Estendidos 1 ). Tal como na filogenia de máxima verossimilhança, o novo genoma antigo, ZH1540, ocupa uma posição basal dentro do clado TEN, com todas as estirpes modernas de TEN formando um subgrupo monofilético (probabilidade posterior 0,96; Fig. 3a ). A maioria das cepas SS14 se enquadra no subclado SS14-Ω previamente definido 4 , 16 , que também recebe alto suporte posterior. De acordo com os resultados das análises de regressão raiz-a-ponta (Extended Data Fig. 4 ), um modelo de relógio relaxado não correlacionado log-normalmente distribuído (UCLD) e um modelo de relógio relaxado não correlacionado distribuído exponencialmente (UCED) foram escolhidos para a análise de datação do relógio molecular, ambos com um prior lognormal estreito com uma média (no espaço real) de 1 × 10 −7 substituições por site por ano e dp de 0,25 na taxa média de clock. Consistente com os resultados relatados anteriormente de datação por relógio molecular 4 , 35 , descobrimos que todas as cepas históricas de TPA são basais a todas as cepas modernas de SS14 e, juntas, formam um clado bem suportado (probabilidade posterior de 0,97). Portanto, consideramos que as cepas históricas se enquadram no clado SS14.
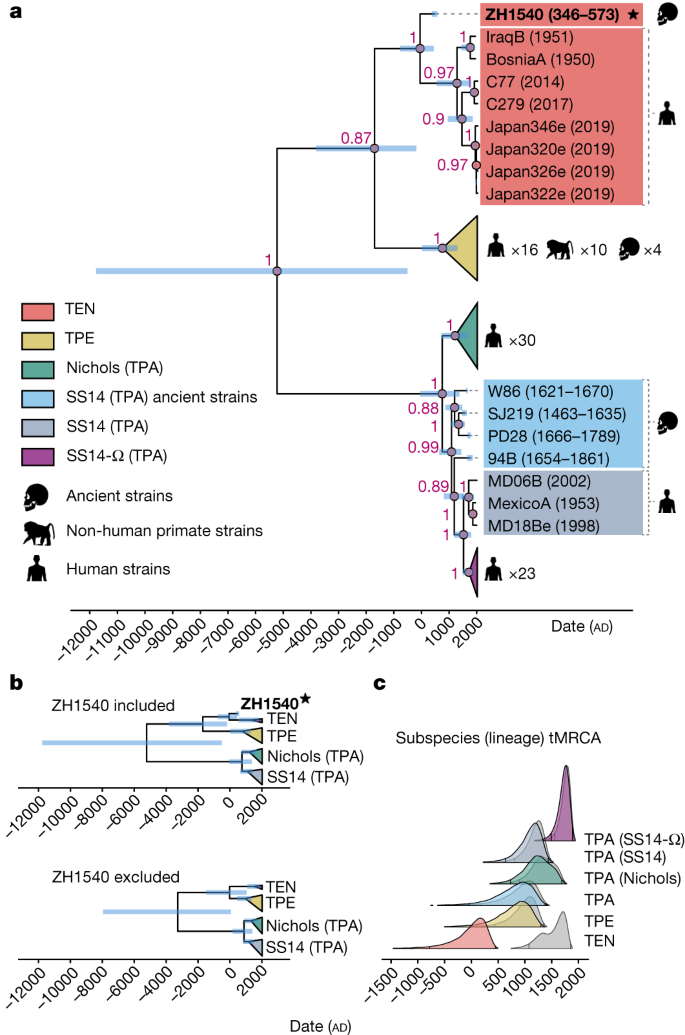
a , árvore de máxima credibilidade do clado (MCC) de genomas antigos e modernos publicados anteriormente, e o genoma antigo ZH1540 deste estudo ( n = 99). As barras azuis indicam os intervalos HPD de 95% das idades dos nós e o texto em vermelho a probabilidade posterior de um grupo ser monofilético (mostrado apenas para nós com probabilidade posterior> 0,8). b , Árvore MCC recolhida simplificada da análise com ZH1540 incluído (parte superior) e excluído (parte inferior). c , Densidades posteriores dos tempos dos tMRCAs da subespécie T. pallidum e linhagens principais estimadas pela datação do relógio molecular com ZH1540 incluída (densidades coloridas, correspondentes a b , superior) e excluídas (densidades cinza, correspondentes a b , inferior ). As linhas verticais dentro das curvas de densidade indicam os limites superior e inferior dos intervalos de 95% HPD.
A idade de ZH1540, que é parametrizada pelos resultados da datação por radiocarbono, atua como uma restrição no tempo do ancestral comum mais recente (tMRCA) do clado TEN. A inclusão de uma amostra muito mais antiga resulta em tempos de divergência anteriores, com intervalos de credibilidade mais amplos para todos os clados principais da árvore (Fig. 3b e Dados Estendidos Figs. 5 – 7 ). Este efeito é mais pronunciado para TEN, onde o intervalo de densidade posterior (HPD) 95% mais alto do tMRCA se estende de 780 aC a 449 dC (236-1845 dC para o subclado compreendendo apenas cepas modernas de TEN), mas é limitado a 1077– de 1855 Anúncio ao excluir ZH1540 (Dados Estendidos Figs. 5 – 7 e Tabela de Dados Estendidos 1 ). Para todas as outras linhagens principais, o efeito é mais moderado e, embora o limite inferior do intervalo HPD de 95% possa ser várias centenas de anos mais antigo ao incluir ZH1540 (cerca de 400 anos no caso de TPE), o limite superior nunca é muito maior. mais de 50 anos (Fig. 3c , Tabela de Dados Estendidos 1 e Dados Estendidos Figs. 6 e 7 ).
Embora as estimativas medianas dos tempos de divergência de linhagem sejam mais antigas do que aquelas relatadas anteriormente (TEN, 47 ad ; TPE, 835 ad ; TPA, 844 ad ; Nichols, 1238 ad ; SS14, 1127 ad ; e SS14-Ω, 1738 ad ), o Os intervalos de HPD de 95% se sobrepõem em grande parte às estimativas relatadas em outros lugares 5 , 35 (Tabela de dados estendida 1 ). As duas exceções são TEN e SS14-Ω, que estimamos ter uma origem possivelmente muito mais antiga do que se pensava anteriormente, independentemente de ZH1540 estar incluído. Isto provavelmente se deve ao conjunto de dados mais diversificado usado aqui, que representa com mais precisão toda a diversidade genética da linhagem SS14-Ω. Da mesma forma, ao incluir ZH1540, o tMRCA geral de T. pallidum é estimado como muito mais antigo do que o estimado anteriormente 4 , 5 , 35 .
Advertimos que, embora tenhamos realizado uma análise de relógio relaxado, não modelamos explicitamente as taxas de substituição específicas da linhagem ou dependentes do tempo. Ambos os fenômenos poderiam explicar a idade mais avançada das linhagens de TPA estimadas aqui em comparação com estudos anteriores, e as taxas dependentes do tempo também poderiam empurrar as subespécies e os tMRCAs gerais de T. pallidum ainda mais para o passado. Como tal, os resultados aqui apresentados devem ser interpretados como limites inferiores nos tempos de divergência dos clados de T. pallidum , deixando aberta a possibilidade de estimar tempos de divergência mais antigos com a recuperação de genomas antigos de maior qualidade e o desenvolvimento de modelos de relógio molecular melhorados. .
Discussão
Muitas hipóteses anteriores – baseadas apenas em evidências paleopatológicas – sugeriram infecções treponêmicas precoces entre as populações pré-históricas nas Américas 2 , 30 , 39 . Aqui apresentamos evidências antigas de DNA de uma treponematose pré-colombiana do Novo Mundo, reconstruindo um genoma de T. pallidum de alta cobertura recuperado de restos humanos indígenas brasileiros de quase 2.000 anos de idade, juntamente com três genomas de baixa cobertura do mesmo contexto espaço-temporal. Inesperadamente, estes genomas são notavelmente semelhantes aos do agente causador do bejel moderno, T. pallidum endemicum . Como a sífilis tem sido o foco central da pesquisa sobre o Treponema , as treponematoses endêmicas têm recebido menos atenção 2 , 40 . Ao contrário da bouba e da sífilis, ambas encontradas anteriormente em contextos do Velho e do Novo Mundo desde o início do período moderno 3 , 4 , 5 , 6 , este genoma recentemente reconstruído representa o primeiro patógeno semelhante ao TEN isolado de vestígios arqueológicos e a única comparação antiga com o conjunto atual de oito genomas de bejel publicados 17 , 38 , 41 , 42 . Casos recentes mostraram que as treponematoses são ocasionalmente transmitidas de forma atípica para as suas subespécies geneticamente confirmadas e desafiaram as suas categorizações geográficas e baseadas em sintomas. 43 , 44 , 45 . Nossas descobertas neste estudo apenas reforçam esta visão: um antigo agente semelhante ao NET, identificado longe do nicho geográfico moderno da doença, em uma região costeira brasileira úmida, atesta a capacidade dos treponemas de se adaptarem a vários climas e localizações geográficas. Excluindo os danos ósseos observados em alguns dos restos estudados, os sintomas clínicos, a gravidade e a história evolutiva do antigo patógeno semelhante ao NET recém-descoberto permanecem desconhecidos. Na verdade, as descobertas de outros patógenos antigos, como a presença da peste na Eurásia desde o final do Neolítico, 46 , e Salmonella paratyphi C, com a possível conexão com a grande epidemia de cocoliztli no México em meados do século XVI, mostraram que doenças historicamente devastadoras podem ter representado sorovares inesperados e exibir hoje uma distribuição muito alterada 47 , 48 , 49 , 50 . O DNA treponêmico de alta qualidade recuperado de uma fonte pré-histórica valida o uso de técnicas antigas de DNA no estabelecimento de uma hipótese inteiramente nova e mais informada sobre os eventos que levaram à disseminação do Treponema pallidum pelo mundo.
Bejel em foco
Atualmente, a doença tropical negligenciada bejel é encontrada principalmente nas regiões áridas da África, da Ásia Ocidental e do Mediterrâneo, tornando-a uma candidata improvável a uma potencial treponematose sul-americana em um contexto costeiro. 23. Although genetically unconfirmed, palaeopathological cases of potential treponematoses found worldwide2,51,52,53 may indicate that bejel was previously more widespread and possibly associated with different environmental habitats. Our genomic investigation, together with the radiocarbon dating of both human remains and stratigraphy, places the newly found treponematosis in South America long before the European contact in the 15th century, even predating the Viking expeditions to the North American coast—firmly attesting to the presence of bejel-like treponemal infections in the pre-contact New World. Phylogenetically, this prehistoric form belongs indisputably to the TEN clade, basal to all of its modern strains. Overall, the TEN genomes are highly similar to each other, which may indicate a slow evolution of the lineage as a whole, at least until recently. Regardless of the improved genomic representation of the modern TEN genomes and the newly reconstructed pre-Columbian genome in this study, a larger representation of this lineage would be needed to draw robust conclusions about the evolution and diversification of the subspecies.
Consequências para do T. pallidum a evolução
Os dados apresentados aqui incluem um genoma treponêmico antigo excepcionalmente de alta qualidade e alta cobertura 4 , 5 , 6 , 35 , e adiar as datas das mais antigas cepas de T. pallidum reconstruídas em mais de 1.000 anos. Embora os antigos fragmentos de DNA recuperados neste estudo não fossem adequados para montagem de novo ou análises pan-genômicas, a alta cobertura obtida permitiu-nos realizar uma análise detalhada ao nível do gene e do SNP. Nossas descobertas revelam numerosos eventos de recombinação inter-subespécies, que são conhecidos por serem um mecanismo chave na evolução bacteriana que resulta, por exemplo, na aquisição de fatores de virulência ou outras características adaptativas. Uma vez que os eventos de recombinação aqui identificados envolvem estirpes antigas e modernas, pelo menos uma das formas endémicas permaneceu em proximidade geográfica e num conjunto de hospedeiros comum com as estirpes de TPA após a sua divergência inicial. Quando e onde exatamente esses eventos de recombinação ocorreram é desconhecido. No entanto, a divergência de clados pode ser estimada através da datação por relógio molecular para os diferentes ramos da árvore filogenética de T. pallidum . A calibração deste método é baseada nas idades conhecidas dos genomas utilizados, tornando genomas antigos datados por radiocarbono com segurança indispensáveis para a análise. Nosso novo genoma antigo de alta cobertura fornece um ponto de calibração pré-histórico sem precedentes para a datação do relógio molecular e nos permite concluir que todas as três subespécies já haviam divergido umas das outras antes das viagens de Colombo. As novas estimativas para o tMRCA de todos T. pallidum (12,006–545 bc) and the emergences of the modern clades (TEN: 780 bc–449 ad, TPE: 28–1299 ad and TPA: 42 bc–1376 ad) are much earlier than previous estimates that relied on modern and previously published historical genomes. Yet, these are only the lower bounds of the divergence times, and the subspecies could have originated even earlier. Only genetically ancestral forms of treponemes could illuminate whether the early American strains spread with the early human dispersals—some 15,000–23,000 years ago29,54—ou resultou de um evento local, talvez zoonótico. Finalmente, como a descoberta revolucionária de uma treponematose pré-colombiana aqui é o resultado de uma combinação de genómica de patógenos antigos e da seleção cuidadosa de amostras arqueológicas, podemos esperar que descobertas futuras iluminem os eventos que levaram ao surgimento e propagação da sífilis venérea, e ajudar a resolver os fatores evolutivos responsáveis pelo sucesso global da Treponema . família
Métodos
Inclusão e ética
Os estudos genéticos de doenças humanas antigas esclarecem como as populações do passado prosperaram e lidaram com problemas de saúde, o que pode desencadear preocupações como a estigmatização devido a doenças ou direitos e questões legais entre as pessoas que vivem hoje. As injustiças históricas, a colonização e a expropriação complicaram frequentemente a capacidade das comunidades indígenas de afirmarem e manterem os seus direitos territoriais num quadro jurídico ou administrativo. É, portanto, crucial considerar, além dos aspectos científicos, também as perspectivas das comunidades e pessoas vivas (indígenas) ao realizar este trabalho. 55 .
Aqui estudamos restos humanos de indivíduos totalmente anônimos que morreram há mais de 1.000 anos e foram sepultados no sítio arqueológico Jabuticabeira II, no município de Tubarão, no estado de Santa Catarina, Brasil. Este local foi escavado por P. de Blasis e equipe 56 , financiado pela Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP). e foi obtida autorização de pesquisa junto ao Instituto do Patrimônio Histórico e Artístico Nacional (IPHAN), conforme correspondência 1793/2019 GAB PRESI-IPHAN do processo 01506.000720/2019-65 de K. Santos Bogia. A utilização das amostras dos restos mortais para este estudo também foi aprovada por P. de Blasis, guardião das coleções Jabuticabeira II do Museu de Arqueologia e Etnologia da Universidade de São Paulo. Os restos mortais foram curados, estudados e amostrados por SE e equipe da Universidade de São Paulo até 2016 e, posteriormente, no Museu de História Natural de Viena.
Os territórios e sítios que abrangem o Rio Grande do Sul, Santa Catarina e Paraná são inerentes à herança ancestral das comunidades Kaingang, Guarani e Xokleng (também chamados de 'povo Sol' ou 'grupo costeiro'), que ainda vivem hoje na região. Estas sociedades não só utilizaram a região para mobilização e migração em busca de abastecimento alimentar, mas também percorreram tradicionalmente distâncias significativas, deixando um rasto de marcas culturais, particularmente no domínio das práticas funerárias. Em um estudo anterior 57, samples of five of the individuals exhumed at Jabuticabeira II were studied, revealing some genetic affinity with the Kaingang (a Ge-speaking group of Southern Brazil). However, to the best of our knowledge, the Kaingang are not seen as direct descendants of sambaqui societies, nor do they identify with the people who once dwelled at Jabuticabeira II or request their remains. Finally, research in the Instituto Socioambiental (https://www.socioambiental.org/; for the defence of Brazilian socio-environmental diversity, including Indigenous Rights) states that the region around Jabuticabeira II is not part of any Indigenous reserve, nor are there claims of groups for territorial rights of this region or for the archaeological remains of this site (P. de Blasis, personal communication).
Os processos degenerativos, muitas vezes resultantes de contextos de marginalização, conflito e deslocamento, testemunham o impacto das relações históricas dos grupos indígenas com os colonizadores e invasores. As aflições e doenças vividas por estes grupos carregam ramificações históricas e ambientais de notável significado, garantindo reconhecimento e exame explícitos. Em relação à possível estigmatização das comunidades locais (indígenas) e das pessoas afetadas pelo bejel, deve-se ressaltar que esta doença contagiosa é uma doença endêmica, principalmente não sexualmente transmissível, comum em regiões quentes onde as pessoas vivem em contato próximo umas com as outras, não têm necessidade de cobrir especialmente roupas e compartilhar utensílios. Hoje, o bejel, que pode levar à estigmatização devido a feridas desfigurantes, ocorre especialmente em comunidades do Mediterrâneo Oriental e da África Ocidental com acesso limitado a cuidados médicos modernos. Embora a Organização Mundial da Saúde perceba a importância das ações tomadas para erradicar o bejel em todo o mundo desde 1949 (WHA2.36 Bejel e outras treponematoses ( https://www.who.int/publications/i/item/wha2.36) ), a doença não é vista como um problema atual de saúde pública no Brasil, como é para alguns outros países 58 , 59 . Isto contrasta com as altas prevalências de doenças sexualmente transmissíveis, como o HIV e a sífilis venérea, que afetam as comunidades indígenas no Brasil (Em São Paulo, ação em aldeias promove debate e testagem rápida de HIV e Sífilis — Fundação Nacional dos Povos Indígenas ( http://www.gov.br/funai/pt-br/assuntos/noticias/2019/em-sao-paulo-acao-em-aldeias-promove-debate-e-testagem-rapida-de-hiv-e- sífilis )). É notável, entretanto, que, no contexto arqueológico, nada implicava que aqueles povos pré-históricos de Jabuticabeira II portadores da treponematose local teriam sido discriminados em sua época e cultura.
Além disso, as descrições culturalmente insensíveis em artigos de investigação paleogenómica são uma questão ética de preocupação 60 . Para garantir a discrição, selecionamos expressões potencialmente insensíveis ou discriminatórias no manuscrito. É importante ressaltar que tivemos a ajuda inestimável de E. Krenak, líder da Cultural Survival no Brasil, ativista indígena e doutorando na Universidade de Viena, para analisar criticamente nossos textos e fornecer conselhos sobre o uso eticamente correto e justo da terminologia.
Informação arqueológica
Os sambaquis da região de Laguna
Um sambaqui é o tipo predominante de sítio arqueológico na costa brasileira: um monturo ou montículo de conchas construído pelo homem, de dimensões variadas, localizado em áreas ricas em recursos, como lagoas, manguezais ou estuários. Sambaquis consistem em sedimentos inorgânicos, conchas de moluscos, restos de alimentos e matéria orgânica misturados em intrincadas estratigrafias associadas a funções domésticas e/ou funerárias. 61 . Mais de 1.000 sambaquis estão mapeados ao longo dos 7.500 km de extensão da costa brasileira e são datados entre 7.500 e 1.000 anos bp 61 , 62 . Pesquisas arqueológicas recentes sugerem que essas populações construtoras de sambaquis eram sedentárias, com uma abundante e estável subsistência marinha, horticultura e alto crescimento populacional. 61 , 63 , rituais funerários elaborados 64 e apropriação da paisagem 56 .
Jabuticabeira II excavation site
Jabuticabeira II (UTM 22 J − 0699479E; 6835488 S) é um sambaqui de médio porte (400 × 250 × 10 m de altura), assentado sobre uma paleoduna e localizado na região de Laguna, área de maior densidade de sambaquis do sul Costa brasileira, a 3 km da Laguna do Camacho, uma das diversas fontes de água associadas a um sistema geológico barreira-lagoa formado durante o Holoceno (Fig. 1 ). Jabuticabeira II, construída ao longo de quase mil anos, é um dos 65 sambaquis mapeados ao redor do sistema lagunar. Este grande número de assentamentos e sua história de ocupação cronologicamente sobreposta atestam uma ocupação bastante densa e intensas interações dos construtores de sambaqui entre 7.500 e 900 anos calibrados (ano cal) bp 56. According to stratigraphic studies, Jabuticabeira II is the result of incremental funerary rituals accumulated over centuries. Although Jabuticabeira II was not completely excavated, 204 burials containing the remains of 282 individuals were exhumed from a 373 m2 area64. Radiocarbon dates of Jabuticabeira II stratigraphy56,64 suggest a long occupation period between 1214–830 cal bc and 118–413 cal ad or 3137–2794 to 1860–1524 cal yr bp (2σ), roughly in line with the radiocarbon datings from bone material of the four individuals in this study, ranging from 350 cal bc to 573 cal ad.
Os restos mortais do sambaqui Jabuticabeira II foram encontrados em sepultamentos simples, duplos e múltiplos, dispersos em aglomerados. Os esqueletos recuperados estavam em sua maioria incompletos, evitando estimativas categóricas de idade e sexo ou outros achados osteológicos. O padrão de sepultamento era bem flexionado e sugeria tratamento intencional do corpo antes do internamento. O pequeno tamanho das sepulturas sugeria que os corpos sofreram dessecação ou decomposição prévia de tecidos moles, mas não o suficiente para produzir desarticulação completa (os ossos das mãos e dos pés foram encontrados articulados). Muitos enterros vêm de perfis e estão incompletos. Os ossos de vários indivíduos estão manchados de ocre vermelho 65 , uma prática comum em sítios arqueológicos do estado de Santa Catarina 66 , 67 . As oferendas são comuns em contextos funerários de sambaqui e incluem adornos confeccionados com material faunístico e ferramentas líticas em diversas formas, desde entulhos até ferramentas polidas e zoólitos, com diferenças na frequência de ocorrência entre diferentes locais e estratos 68 . A oferta mais comum na Jabuticabeira II era o peixe.
Ao todo, 99 indivíduos de Jabuticabeira II, com e sem alterações ósseas sugestivas de infecção, foram triados quanto ao conteúdo de DNA do patógeno. 37 amostras foram consideradas positivas para DNA treponêmico na triagem inicial e quatro amostras produziram dados suficientes para a reconstrução do genoma de T. pallidum (Tabela Suplementar 1 ).
Palaeopathological analysis of treponematoses
Bioarchaeological analyses showed results compatible with increasing population growth and high population density in Jabuticabeira II, including high frequencies of nonspecific stress markers69 and occasional infant stress70, but no evidence of trauma associated with interpersonal conflicts over resources or territory69.
Há, no entanto, evidências de doenças sistêmicas transmissíveis em Jabuticabeira II e outros sambaquis locais brasileiros 30. Eleven 14C accelerator mass spectrometry dates obtained directly from the presumably treponematosis-affected individuals suggest that these diseases are very old on the east coast of South America, with a time-range between 6,300 and 500 yr BP. Among the possible treponemal cases based on osteological analysis, three came from Jabuticabeira II. However, these did not overlap with the individuals yielding the detected genetic evidence in this study.
Informações sobre indivíduos
Indivíduo 41A-L2.05-E4, amostra ZH1390
O indivíduo é um homem adulto de constituição robusta, com estatura estimada em 150,49 ± 2,6 cm (ref. 70 ). Embora fragmentados, os ossos deste indivíduo constituíam um esqueleto quase completo (80%), articulado e enterrado em uma matriz oval rica em conchas em posição hiperflexionada. Os ossos do indivíduo apresentavam sinais de doença infecciosa sistêmica em membros inferiores. Fêmures, tíbias e fíbulas apresentam periostite generalizada discreta e osteoartrose. Também foi observado alargamento na porção lateral das clavículas. Segundo Filippini et al. 30 , aplicando o método ESPIRAL 71 , a doença desse indivíduo poderia ser classificada de forma não conclusiva como sífilis, bouba ou bejel. A amostragem foi realizada em lesão ativa no fragmento da tíbia.
FS9–L3–T2 individual, amostra ZH1540
A amostra vem de um conjunto de ossos misturados, provavelmente de mais de um indivíduo. Os ossos atribuídos a este indivíduo consistem em vários elementos esqueléticos, alguns com alterações patológicas, como osteomielite grave no terço distal do úmero direito, periostite grave na ulna esquerda, periostite na diáfise da fíbula e dois corpos vertebrais com osteofitose. A amostra foi retirada do fragmento da fíbula, na área com periostite.
FS3B-L3-T4 individual, amostra ZH1541
A amostra vem de um de três indivíduos separados, encontrados misturados. Os elementos esqueléticos pertencentes a este adulto robusto de idade e sexo desconhecidos incluem um rádio esquerdo com artrite, um fragmento da ulna esquerda (muito robusto), um fragmento do úmero esquerdo, fragmentos de um fêmur, uma tíbia e uma fíbula e um primeiro metatarso. A amostra foi retirada de um fragmento de fêmur, sob a superfície imediata do osso, para melhor evitar a possível introdução de contaminantes externos.
Individual 2B-L6-E3, ZH1557
A amostra provém de um indivíduo provavelmente adulto do sexo masculino. O indivíduo estava articulado e em posição flexionada com outro indivíduo adulto do sexo feminino enterrado em cima. Os achados osteopatológicos nos ossos do indivíduo amostrado incluíram sinais de doença articular degenerativa, osteoartrite intervertebral lombar grave, escoliose e possíveis lesões na patela. No entanto, não foram observadas lesões típicas sugestivas de infecção treponêmica. A amostra foi retirada de um pequeno pedaço de osso longo, sob a superfície imediata do osso, para melhor evitar a possível introdução de contaminantes externos.
Correção do efeito do reservatório marinho para 14 C namoro
A datação por radiocarbono foi realizada pelo Laboratório de Física de Feixes de Íons da ETH Zurich (laboratório número: ETH-127328) utilizando colágeno ósseo purificado por um método de ultrafiltração modificado 72 . A calibração dos dados foi feita com OxCal v4.4.4. A dieta dos habitantes de Jabuticabeira II, constituída substancialmente por fontes alimentares marinhas, produz efeito reservatório nas datas de radiocarbono calculadas como idade média de 247,8 ( σ = 103,7) anos 73 . Considerando a alta contribuição de carbono marinho para o colágeno ósseo dos indivíduos de Jabuticabeira II, as datas de radiocarbono foram modeladas com Calib Rev 8.20 74 ( http://calib.org/calib/calib.htm ) usando a curva de calibração Mixed Marine SHCal20 75 , 76 e aplicando o valor médio estimado de correção de reservatórios de radiocarbono marinhos locais (Δ R ) de -126 ± 29 para a costa sul do Brasil (banco de dados de Correção de Reservatórios Marinhos) 73,77. We considered the average relative contribution of marine carbon to collagen derived from Bayesian Mixing Models for Jabuticabeira II individuals, calculated at a mean value of 42.5%78,79. Para as estimativas individuais para as amostras, consulte a Tabela Suplementar 2 .
Sample processing
Samples were documented and carried through sampling, DNA extraction, library preparation and library indexing in facilities dedicated to ancient DNA work at the University of Zurich, including decontamination of samples, laboratory equipment and reagents with UV irradiation and using protective clothing and minimum contamination-risk working methods.
Todas as etapas pós-amplificação foram realizadas nas instalações laboratoriais regulares disponíveis para o Grupo de Paleogenética do Instituto de Medicina Evolutiva (IEM) da Universidade de Zurique (UZH). O sequenciamento de DNA foi realizado nas instalações de Sequenciamento de Próxima Geração das Instalações Principais do BioCenter de Viena (VBCF) ou no Centro de Genômica Funcional da Universidade de Zurique (FGCZ).
Extração de DNA antigo
Todas as superfícies das amostras foram irradiadas com luz ultravioleta para minimizar a contaminação potencial do DNA moderno. O pó ósseo foi obtido utilizando-se broca odontológica e brocas com cabeça diamantada. A extração de DNA foi realizada em cerca de 50-100 mg de pó de osso, de acordo com um protocolo de extração bem estabelecido para DNA antigo. 80 . Os controles negativos para processos de extração e biblioteca foram processados paralelamente em todos os experimentos, um controle para cada dez amostras, sequenciados e comparados bioinformaticamente com seus correspondentes lotes de amostras, como precaução contra possível contaminação.
Preparação da biblioteca
Bibliotecas de DNA de cadeia dupla foram produzidas para triagem inicial com sequenciamento shotgun, sem tratamento UDG (ou seja, tratamento químico com o objetivo de limitar os danos relacionados à idade no DNA). Duas bibliotecas adicionais para cada uma das amostras potencialmente positivas da primeira rodada de captura foram produzidas para maximizar a complexidade do DNA. Para a preparação de bibliotecas de DNA, 20 µl de extrato de DNA foram convertidos em bibliotecas de DNA de fita dupla 31 . Códigos de barras (índices) específicos da amostra foram adicionados a ambas as extremidades dos fragmentos de DNA nas bibliotecas 81 . As bibliotecas indexadas foram então amplificadas para atingir uma concentração mínima de DNA de aproximadamente 90 ng ml −1 . A amplificação foi realizada utilizando 1 × tampão Herculase II, 0,4 mM IS5 e 0,4 mM IS6 primer 81 , DNA polimerase de fusão Herculase II (Agilent Technologies), dNTPs 0,25 mM (100 mM; 25 mM cada dNTP) e 5 ml de biblioteca indexada como modelo de DNA. Foram preparadas quatro reações por biblioteca e o volume total da reação de amplificação foi de 100 ml. O perfil térmico incluiu uma desnaturação inicial por 2 min a 95 °C e 3-18 ciclos, dependendo da concentração de DNA após a indexação das bibliotecas, desnaturação por 30 s a 95 °C, 30 s de recozimento a 60 °C e 30 s de recozimento a 60 °C. alongamento a 72°C, seguido por uma etapa de alongamento final por 5 min a 72°C. Todas as divisões de uma biblioteca indexada foram reunidas e purificadas utilizando o kit de purificação QIAGEN MinElute PCR. As bibliotecas de DNA foram então quantificadas com D1000 ScreenTape em um Agilent 2200 TapeStation (Agilent Technologies) e combinadas em pools equimolares para sequenciamento.
Pathogen screening
Os dados do Shotgun foram usados para uma triagem inicial das 99 amostras candidatas, com o software Kraken2 82 , e 41 amostras que tiveram mais de 7 ocorrências para T. pallidum foram selecionadas para enriquecimento alvo. As amostras selecionadas foram submetidas a um processo de enriquecimento alvo e posteriormente processadas pelo FastQ Screen v0.15.1 83 para verificar o número de leituras mapeadas em relação a três genomas de referência representativos de alta qualidade da subespécie de T. pallidum (CDC2, BosniaA e Nichols). As nove amostras mais promissoras (> 5.000 acessos de Kraken a T. pallidum após a primeira rodada de captura em solução) foram transformadas em duas bibliotecas extras e recapturadas conforme explicado em detalhes nas seções a seguir.
Enriquecimento alvo para T. pallidum DNA de
O enriquecimento de bibliotecas de cadeia dupla em todo o genoma foi realizado através de kits de enriquecimento de alvo personalizados (Arbor Bioscience). Iscas de RNA com comprimento de 60 nucleotídeos e densidade de ladrilhos de 4 pb foram projetadas com base em três genomas de referência: Nichols ( CP004010.2 ), SS14 ( CP000805.1 ), Fribourg-Blanc ( CP003902 ). Conjuntos de bibliotecas de 500 ng foram enriquecidos de acordo com as instruções do fabricante. As bibliotecas capturadas foram amplificadas em reações de 100 µl contendo 1 unidade de DNA polimerase de fusão Herculase II (Agilent), 1 × tampão de reação Herculase II, dNTPs 0,25 mM, iniciadores IS5 e IS6 0,4 mM. 81 e modelo de biblioteca de 15 µl, com o seguinte perfil térmico: desnaturação inicial a 95 °C por 2 min, 14 ciclos de desnaturação a 95 °C por 30 s, recozimento a 60 °C por 30 s e alongamento a 72 °C por 30 s, seguido de um alongamento final a 72°C por 5 min. As bibliotecas capturadas foram purificadas com colunas de rotação MinElute (QIAGEN) e quantificadas com uma ScreenTape de alta sensibilidade D1000 em um Agilent 2200 TapeStation.
Sequencing
For both shotgun data retrieval and after the capture processing, the samples were pooled in unimolar quantity (for SG sequencing up to 50 samples per pool, and for the capture process 2–8 samples per pool), and sequenced on an Illumina NextSeq500 with 2 × 75 + 8 + 8 cycles using the manufacturer’s protocols for multiplex sequencing at the Functional Genomics Center in Zurich or at the Vienna BioCenter Core Facilities.
Statistical analyses
Dataset selection
Montamos um conjunto de dados genômicos compreendendo 98 genomas de T. pallidum disponíveis publicamente (8 TEN, 30 TPE e 60 TPA) de estudos publicados anteriormente (incluindo 8 genomas antigos) e o recém-gerado genoma ZH1540. Os genomas representam a variação genética das três subespécies conhecidas de T. pallidum (TPA, TPE e TEN) disponíveis até dezembro de 2022, e foram selecionados com foco em TEN e TPE, devido à sua proximidade com o novo genoma antigo classificado como TEN .
Os dados publicados para o conjunto de dados do genoma moderno neste estudo estão disponíveis no banco de dados European Nucleotide Archive (ENA): PRJNA313497 (números de acesso: SRR3268682 , SRR3268724 , SRR3268715 , SRR3268694 , SRR3268696 , SRR3268709 , SRR3268710 ), 11481 (números de acesso: ERR1470343 , ERR3596780 , ERR3596747 , ERR3596783 ), PRJEB28546 (números de acesso: ERR4045394 , ERR3684452 , ERR3684456 , ERR3684465 , SRR13721290 , ERR4853530 , ERR4993349 , ERR485358 7 , ERR4899206 , ERR5207017 , ERR5207018 , ERR5207019 , ERR4899215 , ERR4853623 , ERR4853625 ), PRJNA508872 (números de acesso: SRR8501165 , SRR8501164 , SRR8501167 , SRR8501166 , SRR8501168 , SRR8501171 ), PRJNA723099 (números de acesso: SRR14277267 , SRR14277266 , SRR14277458 , SRR14277444 ), PRJEB11481 (número de acesso: ERR 1470331 ), PRJDB9408 (números de acesso: DRR213712 , DRR213718 ), PRJNA588802 (números de acesso: SRR10430858 , SRS5636328 ), PRJNA322283 (número de acesso: SRR3584843 ), PRJNA754263 (números de acesso: SRR15440297 , SRR15440150 , SRR15440451 , SRR15440240 ), PRJEB40752 (números de acesso: ERR4690809 , ERR4690806 , ERR4690810 , ERR4690812 , ERR4690811 ). Arquivos de montagem foram usados para 9 genomas do banco de dados do National Center for Biotechnology Information (NCBI): CP002375.1 , CP002376.1 , NC_016842.1 , NC_017268.1 , NC_018722.1 , NC_021490.2 , NC_021508.1 , GCA_000813285.1 , CP035193.1 e para 24 genomas modernos do European Nucleotide Archive (ENA): CP021113.1 , CP073572.1 , CP073557.1 , CP073553.1 , CP073536.1 , CP073526.1 , CP073490.1 , CP073487.1 , CP07347 0 .1 , CP073447.1 , CP073446.1 , CP073399.1 , CP040555.1 , LT986433.1 , LT986434.1 , CP032303.1 , CP020366.1 , CP024088.1 , CP024089.1 , CP07812 , 1.1 , CP078090.1 , CP081507.1 , CP051889.1 e CP003902 .1 . Os dados de sequência bruta (arquivos fastq) usados para 6 genomas modernos estão disponíveis no banco de dados NCBI: PRJEB20795 (números de acesso: ERS1724928 , ERS1724930 , ERS1884567 ) e PRJNA343706 (números de acesso: SRR4308604 , SRR4308606 , SRR4308597 ). Genomas treponêmicos antigos publicados anteriormente aqui usados estão disponíveis na ENA: PRJEB37490 (número de acesso: ERR4065503 ), PRJEB37633 (número de acesso: ERR4000645 ), PRJEB35855 , PRJEB21276 (números de acesso: ERS2470995 , ERS2470994 ) e PRJEB62102 . Informações detalhadas sobre a fonte do conjunto de dados de referência estão documentadas na Tabela Suplementar 3 .
Selecionamos todos os oito genomas TEN disponíveis publicamente, todos com mais de 99,4% de cobertura genômica, com exceção de C77 17 (81,4%). Selecionamos 30 genomas de TPE (Tabela Suplementar 3 ). Para representar cada linhagem ou sub-linhagem, selecionamos pelo menos um genoma, preferindo aqueles com maior profundidade de sequenciamento e cobertura genômica. Todos os genomas TPE incluídos têm mais de 95,3% de cobertura genômica, exceto os quatro antigos genomas TPE: SJN003, AGU007, 133 e CHS119, exibindo 97,4%, 92,7%, 57% e 62% de cobertura genômica, respectivamente. Além disso, foram incluídos 60 genomas de TPA das principais linhagens e sub-linhagens descritas em estudos anteriores (Tabela Suplementar 3 ). Todos esses genomas tinham mais de 90% de cobertura, exceto os quatro genomas antigos, PD28, W86, SJ219 e 94B, todos com cobertura genômica de 30% ou mais. Todos os genomas no conjunto de dados são separados uns dos outros por pelo menos 5 SNPs. A cepa TPA Seattle-81 foi excluída do conjunto de dados final devido a mutações provavelmente acumuladas durante passagens extensas em coelhos que podem causar posicionamento ambíguo em filogenias 4 , 16 , 36 .
Os dados brutos e/ou arquivos de montagem para cada genoma em nosso conjunto de dados foram baixados dos bancos de dados públicos: European Nucleotide Archive (ENA) 84 e Centro Nacional de Informações sobre Biotecnologia (NCBI) 85 . Os números de acesso são fornecidos na Tabela Suplementar 3 .
Processamento de leitura e geração de alinhamento de genoma baseado em múltiplas referências
To reconstruct the individual genomes from the raw data, we carried out raw read quality control and preprocessing, removing duplicates, variant calling and filtering using the default parameters when not otherwise specified. After processing the de-multiplexed sequencing reads, sample sequencing quality was analysed with FastQC version 0.11.983, filtering reads with a QC value < 25. Following processing by cutadapt version 4.186 to remove the sequencing adapters, in order to reduce the reference bias, and improve the posterior phylogenetic inference and assignment87, the genome reference selection for mapping each sample was determined according to the results from the original manuscript where the genomes were published (see Supplementary Table 3). The mapping was carried out by BWA mem88(usando parâmetros: -k 19, -r 2,5). Foram utilizados quatro genomas de referência; os bem estudados genomas TEN e TPE BosniaA (NZ_CP007548.1) e CDC2 ( NC_016848.1 ), bem como os genomas Nichols ( NC_021490.2 ) e SS14 ( NC_010741.1 ), representando as duas linhagens principais dentro do TPA. No entanto, para as novas amostras antigas obtidas aqui, os genomas de cada amostra foram reconstruídos mapeando três genomas de referência de alta qualidade, representando as três subespécies de T. pallidum (CDC2, BosniaA e Nichols).
CleanSam, do Picard Toolkit versão 2.18.29 ( http://broadinstitute.github.io/picard ), foi usado para limpar os arquivos SAM ou BAM fornecidos. As leituras duplicadas foram removidas usando MarkDuplicates, do kit de ferramentas Picard versão 2.18.29. AddOrReplaceReadGroups, do Picard Toolkit versão 2.18.29, foi usado para atribuir todas as leituras em um arquivo a um único novo grupo de leitura antes de usar mapDamage versão 2.2.0-86-g81d0aca 89 para estimar os parâmetros de dano ao DNA e redimensionar os índices de qualidade de posições provavelmente danificadas nas leituras (usando o parâmetro: --rescale).
Depois de gerar uma saída de pilha de texto para os arquivos BAM com a ferramenta mpileup do Samtools versão 1.7 90 , os SNPs foram chamados usando o VarScan versão 2.4.3 91 (usando parâmetros: -p-valor 0,01, -min-reads2 1, -min-coverage 1, -min-freq-for-hom, 0,4 -min-var-freq 0,05, -output-vcf 1). Em seguida, uma filtragem SNP também foi realizada com VarScan (usando para as amostras modernas os parâmetros: -p-value 0,01, -min-reads2, 5 -min-coverage 10, -min-avg-qual 30 -min-freq-for -hom 0,4, -min-var-freq 0,9, -output-vcf 1 e modificação de alguns parâmetros para as amostras antigas devido à sua menor cobertura e qualidade de leitura: -p-value 0,01 -min-reads2 3, -min-coverage; 5, -min-avg-qual 30, -min-freq-for-hom 0,4, -min-var-freq 0,9 -output-vcf 1). Além disso, todas as posições com menos de 3 leituras mapeadas foram mascaradas com Genomecov do Bedtools versão 2.26.0 92 para amostras modernas e antigas. Todas as etapas de geração do genoma foram visualizadas e confirmadas manualmente com Tablet versão 1.21.02.08 93 , verificando cada SNP um por um e descartando os possíveis SNPs espúrios do novo genoma antigo ZH1540. As sequências finais resultantes foram obtidas por maskfasta do Bedtools v2.26.0.
Além disso, utilizamos metodologias testadas de sequenciamento e análise posterior 17 , 42 para obter maior cobertura e genomas modernos de T. pallidum mais confiáveis . Sempre que possível, foram obtidos arquivos de montagem em vez de dados brutos (Tabela Suplementar 3 ). Um alinhamento de genoma baseado em referência múltipla para todas as sequências foi gerado no MAFFT v7.467 94 (usando parâmetros: --adjustdirection --auto --fastaout --reorder). Porém, devido ao uso de diferentes referências genômicas, regiões com baixa cobertura para alguns genomas, correspondendo principalmente aos genes tpr e arp , foram revisadas e alinhadas manualmente com o Aliview versão 1.25. 95 .
As amostras ZH1390, ZH1541 e ZH1557 tinham dados suficientes para tentar uma reconstrução do genoma e foram determinadas como tendo o maior número de SNPs em comum com a referência TEN, mas foram excluídas das análises downstream devido à cobertura limitada adquirida para cada uma delas, o que fez os SNPs obtidos são menos confiáveis. A amostra ZH1540, no entanto, rendeu uma cobertura genômica notável de 33,6× e foi selecionada para análises aprofundadas subsequentes.
Proteinorto versão 6.0b 96 (usando parâmetros: -p=blastn -singles -keep) foi usado para conduzir um estudo de ortologia a fim de encontrar genes ortólogos nos quatro genomas de referência utilizados 96 . Cada gene presente em pelo menos um dos quatro genomas de referência teve suas coordenadas genômicas determinadas com base em sua localização no alinhamento final mesclado (ver Tabela Suplementar 3 ).
Para verificar a precisão do alinhamento final do genoma múltiplo, e que nenhum gene codificador de proteína foi inadvertidamente truncado, as traduções de proteínas para cada gene presente em pelo menos um genoma de referência foram comparadas com os arquivos gff3 originais de cada uma das quatro referências (Suplemento Tabela 3 ). O genoma ZH1540 reconstruído e suas principais características foram representados graficamente usando BRIG versão 0.95-dev.0003 97 .
Análise de recombinação usando PIM
Como observado anteriormente 36 , a presença de recombinação nos genomas de T. pallidum pode interferir nas topologias das árvores filogenéticas inferidas. Para analisar a potencial recombinação genética, usamos o pipeline PIM 36 para detectar recombinação gene por gene. Em resumo, o processo envolveu as seguintes etapas:
-
(1)
Using IQ-TREE version 1.6.10, a maximum-likelihood tree was created for the multiple genome alignment98. All maximum-likelihood trees for the remaining steps were obtained using GTR99 + G100 + I101 as an evolutionary model and 1,000 bootstraps replications.
-
(2)
The 1,161 genes found in at least one of the reference genomes were extracted, and the number of SNPs for each gene was calculated. Genes with less than three SNPs were excluded.
-
(3)
The phylogenetic signal in each gene alignment for each of the remaining genes was evaluated by likelihood mapping102 in IQ-TREE (using parameters: -lmap 10000 -n 0), retaining only those genes that showed a phylogenetic signal.
-
(4)
A maximum-likelihood tree was generated for each of the remaining genes using IQ-TREE.
-
(5)
Para cada gene incluído, testamos a congruência filogenética entre árvores usando IQ-TREE (usando parâmetros: -m GTR + G8 -zb 10000 -zw), comparando a árvore de máxima verossimilhança obtida a partir do alinhamento do gene e a árvore de máxima verossimilhança obtida do alinhamento do genoma completo usando dois métodos diferentes: Shimodaira – Hasegawa 103 e pesos de verossimilhança esperados (ELW) 104 . Genes para os quais pelo menos um teste rejeitou a topologia da árvore de referência com o alinhamento do gene adotando uma abordagem conservadora ( P <0,2, valor de peso próximo a 0, para os testes Shimodaira-Hasegawa e ELW, respectivamente) e o alinhamento completo do genoma rejeitou a topologia de as árvores construídas utilizando o alinhamento gênico (incongruência recíproca, P < 0,2 e valor de peso próximo a 0) em pelo menos um deles foram selecionadas e examinadas mais de perto na etapa seguinte.
-
(6)
Usando MEGAX 105 , os genes selecionados que apresentavam incongruência recíproca foram posteriormente examinados para avaliar e descrever potenciais eventos de recombinação. Um gene deve ter pelo menos três SNPs homoplásicos próximos – SNPs que são compartilhados por vários grupos (TPE, TEN, TPA-Nichols ou TPA-SS14) e produzir uma distribuição polifilética – para ser rotulado como recombinante. Os SNPs homoplásicos encontrados no alinhamento dos genes serviram como limites das áreas recombinantes.
-
(7)
Utilizando um critério de parcimônia na distribuição de estados alternativos dos SNPs homoplásticos, foram inferidos os potenciais clados ou cepas doadoras e receptoras de cada evento de recombinação.
DNA sections, a number of genes have a high percentage of sites with missing data. The majority of these genes are members of the tpr and arp families, which include collections of paralogous genes. In order to continue analysing these intriguing genes with the PIM pipeline, strains that had a high percentage of missing data in each of these genes were eliminated. Following previous findings35,36, the hypervariable gene tprK (tp0897), with seven hypervariable regions that undergo intrastrain gene conversion17,37,106,107,108,109, and the tp0316 and tp0317 genes, also under gene conversion, were completely excluded from the recombination analysis.
PIM procedure for likelihood mapping and topology tests
Um teste de mapeamento de verossimilhança foi realizado usando IQ-TREE para determinar quais genes (Tabela Suplementar 4 ) mostraram um sinal filogenético (dos 382 genes para os quais> 3 SNPs foram encontrados em comparação pareada com pelo menos um genoma de referência). Para cada quarteto (subconjunto de quatro sequências) nos dados, o teste cria árvores filogenéticas não enraizadas. As probabilidades do quarteto são então traçadas dentro de um triângulo, onde a posição denota a “semelhança em árvore” do quarteto em questão. Os quartetos de canto estão completamente resolvidos, os quartetos nas laterais estão parcialmente resolvidos e os quartetos no centro não estão resolvidos. Dos 382 genes, 29 tinham muitos valores faltantes para serem testados usando o método de mapeamento de verossimilhança. Para incluir esses genes nas próximas etapas do pipeline PIM e nas comparações de topologia, as sequências problemáticas com mais de 50% de posições com dados faltantes foram removidas.
Após o teste de mapeamento de verossimilhança, 9 genes pertencentes à zona central do triângulo foram descartados (Tabela Suplementar 4 ). Em seguida, usando os testes de topologia Shimodaira-Hasegawa e ELW, comparamos as árvores gênicas dos genes restantes com a árvore de referência preliminar do alinhamento do genoma completo para avaliar sua congruência filogenética (Tabela Suplementar 4 ). Dos 373 genes que testaram positivo para incongruência filogenética, 27 continham pelo menos três SNPs consecutivos, apoiando um evento de recombinação. A estes adicionamos tp0859 , que foi detectado como recombinante em um estudo anterior 35 , resultando em um total de 27 genes recombinantes.
Análise de recombinação usando Gubbins e ClonalFrameML
Gubbins version 2.3.1110 and ClonalFrameML version 1.11-1111 are frequently used tools for the genome-wide identification of recombinant positions in bacterial genomes. To test the robustness of our recombination analysis using PIM, we also ran these two programs, with default parameters and the same whole-genome alignment used with PIM. Gubbins identified 301 distinct recombination events associated with 103 genes, ranging in size from 5 bp to 13,866 bp. Similarly, ClonalFrameML detected 656 events, with 32 of them being 1 or 2 bp long, and the longest event spanning 782 bp. Notably, all the genes identified by PIM as having a recombinant region were also detected by both ClonalFrameML and Gubbins, except for gene tp0558, which was missed by ClonalFrameML but detected by Gubbins. Additionally, genes tp0164 and tp0179 were detected by ClonalFrameML but missed by Gubbins.
Phylogenetic analysis
Uma árvore de máxima verossimilhança baseada no alinhamento incluindo todos os genes foi construída com IQ-TREE, usando GTR + G + I como modelo evolutivo e 1.000 replicações de bootstrap (Extended Data Fig. 2a ). Em seguida, os genes identificados como recombinantes pelo PIM foram removidos do alinhamento do genoma múltiplo. Três genes adicionais ( tp0897 , tp0316 e tp0317 ), que contêm regiões repetitivas e foram identificados como hipervariáveis e/ou sob conversão gênica no passado, também foram removidos para evitar a introdução de um viés potencial. Como o gene tp0317 está aninhado dentro do gene tp0316 e as coordenadas do genoma de referência da BósniaA para tp0316 cobriram uma área maior do que as dos outros genomas de referência, tp0316 e tp0317 foram removidos de acordo com as coordenadas tp0316 do genoma de referência da BósniaA. Uma árvore filogenética de referência foi então construída empregando o novo alinhamento do genoma de herança vertical, também com IQ-TREE usando GTR + G + I como modelo evolutivo e 1.000 replicações de bootstrap (Extended Data Fig. 2b ). Ambas as árvores obtidas foram comparadas e são mostradas na Fig. 2 de dados estendidos .
A linhagem SS14 foi descrita anteriormente como um grupo amplamente epidêmico e resistente a macrolídeos que surgiu após, e possivelmente foi motivado pelo, uso clínico de antibióticos após sua descoberta. 12 , 16 . Com base nos resultados da nossa análise filogenética e expandindo as classificações filogenéticas anteriores e a nomenclatura da linhagem SS14 12 , 16 , definimos o clado que contém quase todos os genomas SS14 de amostras clínicas e contemporâneas como a sub-linhagem SS14-Ω. No entanto, duas amostras clínicas contemporâneas (MD18Be e MD06B) não foram classificadas como sub-linhagem SS14-Ω, porque essas amostras se agrupam com o genoma do MéxicoA, em linha com resultados publicados anteriormente 42.
To compare the PIM-based analysis with other widely used recombination detection methods, Gubbins and ClonalFrameML, we followed a similar procedure of removing the recombinant positions detected by these tools and inferred maximum-likelihood trees with the retained positions in the corresponding multiple genome alignments. All the phylogenetic trees with recombination events removed exhibit general congruence with each other, whether the events were identified by PIM, Gubbins or ClonalFrameML. Furthermore, the placement of the ZH1540 genome remained consistent in the phylogenetic trees, regardless of the recombination detection method employed, and despite the elimination of recombinant genes to generate the vertically inherited alignment.
Exploratory characterization of the 16S-23S genes
T. pallidum contains two rRNA (rrn) operons, each of which encodes the 16S-23S-5S rRNA genes and intergenic spacer regions (ISRs). There is evidence that the random distribution of rrn spacer patterns in T. pallidum may be generated by reciprocal translocation of rrn operons mediated by a recBCD-like system found in the intergenic spacer regions (ISRs)112 . De acordo com estudos anteriores 112 , 113 , 114 , 115 , descobrimos que os ISRs 16S-23S das cepas de TPA contêm os genes tRNA-Ile (tRNA-Ile-1; tp0012 ) e tRNA-Ala (tRNA-Ala-3; tp00t15 ) dentro dos rrn1 e rrn2 operons , respectivamente . Por outro lado, os genomas TPE mostram um padrão espaçador Ala/Ile, onde os tp0012 e tp00t15 ortólogos estão localizados dentro dos operons rrn2 e rrn1 , respectivamente.
Identificamos 68 SNPs nos genes r0001 , r0002 , r0004 e r0005 , codificando os genes 16S-23S rRNA do novo e antigo genoma ZH1540, colocando-os entre os genes mais variáveis em nosso alinhamento e aumentando o potencial de que incluí-los no alinhamento poderia resultar em uma reconstrução filogenética tendenciosa. Embora os SNPs encontrados pareçam ser bem suportados pelas leituras obtidas a partir do mapeamento de sequências (Tabela Suplementar 3 ), a sua origem de possível contaminação não pode ser completamente descartada e seriam necessárias análises adicionais para os confirmar.
A exclusão destes genes do alinhamento, além dos genes recombinantes e tp0316, tp0317 e tp0897 , não resultou em quaisquer alterações na topologia (Extended Data Figs. 2 b e 3 ), embora os comprimentos dos ramos tenham sido alterados. Como se sabe que esses genes possuem regiões conservadas, além de regiões variáveis, usadas para explorar as relações evolutivas entre bactérias patogênicas 116 , 117 , 118 , decidimos mantê-los no alinhamento para todas as análises subsequentes. Finalmente, notamos que o genoma ZH1540 não possuía nenhuma das duas mutações do gene de RNA ribossômico 23 S de T. pallidum conhecidas por conferir resistência a macrólidos (A2058G e A2069G). Em contraste, quatro estirpes modernas de TEN do Japão possuem a mutação A2048G, sugerindo uma recente pressão de selecção para mutações de resistência a antibióticos.
Molecular clock dating
We used the Bayesian phylogenetics package BEAST2 v2.6.7119 to estimate a time-calibrated phylogeny of the context dataset of 98 T. pallidum genomes along with our new ancient genome, ZH1540. We removed hypervariable and recombining genes from the alignment, as described above, reduced it to variable sites and used an ascertainment bias correction to account for constant sites.
Análises de regressão da raiz às pontas (Dados Estendidos Fig. 4 ) mostram que, embora haja uma correlação positiva entre o ano de amostragem e a divergência da raiz às pontas entre todas as cepas clínicas modernas, indicando uma evolução semelhante a um relógio, a correlação é muito fraca quando incluindo também cepas passadas e negativo quando incluindo cepas antigas. Dentro dos clados TPE, TEN e SS14 existe uma correlação positiva entre todas as cepas clínicas modernas e passadas. Por outro lado, a correlação é negativa para cepas de Nichols, mesmo quando se olha apenas para cepas clínicas. A fim de levar em conta a variação da taxa e os longos ramos terminais em algumas cepas (provavelmente devido a uma infinidade de efeitos, incluindo erros de sequenciamento, contaminação e mutações introduzidas durante a passagem do coelho), usamos um modelo de relógio UCLD e UCED para a datação do relógio molecular. análise 120 . Para ambos os modelos, colocamos um prior lognormal estreito com uma média (no espaço real) de 1 × 10 −7 substituições por local por ano e desvio padrão de 0,25 na frequência média do clock. Este forte anterior foi usado para compensar o fraco sinal temporal entre os genomas de T. pallidum e foi calibrado em estimativas anteriores da taxa de substituição 4 , 35 . Usamos ainda um modelo de substituição GTR + G + I 118 e um gráfico do horizonte bayesiano 121 modelo demográfico (árvore anterior) com 10 grupos. Para todos os genomas onde as datas de amostragem não são conhecidas exatamente, usamos anteriores uniformes ao longo dos intervalos de datas relatados nos estudos originais para levar em conta a incerteza 4 , 5 , 6 , 16 , 122 . Para ZH1540, definimos o intervalo de datas para 364–573 dC , de acordo com os resultados da datação por radiocarbono corrigidos pelo efeito do reservatório marinho acima. Priores padrão foram usados para todos os outros parâmetros do modelo. A mesma análise foi repetida sem ZH1540, a fim de avaliar o efeito do nosso novo genoma antigo nas datas de divergência. Repetimos ainda mais a análise usando um lognormal anterior amplo com uma média (no espaço real) de 1 × 10 −7 substitutions per site per year and standard deviation 1 on the mean clock rate and using both constant-size and exponential growth coalescent models to assess the impacts of the mean clock rate prior and demographic models on divergence time estimates.
Para cada análise, executamos quatro cadeias de Markov Monte Carlo (MCMC) de 5 × 10 8 steps each, sampling parameters and trees every 10,000 steps. After assessing convergence in Tracer v1.7123 and confirming that all four chains converged to the same posterior distribution, we combined the chains after discarding the first 10% of samples as burn-in. In the resulting combined chains all parameters have effective sample size (ESS) values > 150. TreeAnnotator v2.6.7 was used to compute MCC trees and the results were visualized using ggplot2124, ggtree125 and custom scripts. The 95% HPD of the coefficient of variation estimated under the UCLD model excluded 0 (median = 1.46, 95% HPD 1.08–1.9), indicating that a strict clock model is not appropriate for our dataset. Robustness analyses show that under a narrow mean clock rate prior both the UCED and UCLD clock models result in similar divergence time estimates (Extended Data Fig. 5a–f), with the UCED model estimates tending to be more recent and the UCLD model estimates usually having longer tails. Under a wide mean clock rate prior, estimates with the UCED are broadly similar, albeit wider, while the UCLD model estimates very wide posterior distributions for divergence times, indicating little information under this model. Divergence time estimates were not sensitive to the demographic model used. The MCC trees under the UCED model with a narrow prior, both with and without ZH1540 included in the analysis are shown in Extended Data Figs. 6 and 7, respectively.
Finalmente, realizamos um teste bayesiano de randomização de datas 126 , 127 , 128 (DRT) para avaliar melhor a força do sinal temporal em nosso conjunto de dados, permutando datas de amostragem entre genomas e realizando 50 análises replicadas. Para as análises, utilizou-se o conjunto de dados completo, um modelo de relógio UCED com um anterior estreito e o modelo demográfico Bayesiano do horizonte, fixando as datas de amostragem de cepas antigas às médias dos intervalos de datas de radiocarbono para simplificar. Cadeias MCMC foram executadas por 1 × 10 8 passos, amostragem de parâmetros a cada 10.000 passos. A convergência foi avaliada usando a coda 129 pacote para garantir que todos os parâmetros em todas as cadeias tenham valores de ESS > 150. Os resultados do DRT mostram que os intervalos HPD de 95% da taxa de clock média em réplicas com datas de amostragem permutadas são muito menores do que o esperado se todas as informações vierem da taxa de clock média anterior (Dados Estendidos Fig. 5g ). Em geral, os intervalos HPD não se sobrepõem ao intervalo HPD de 95% da taxa média de clock estimada com as datas de amostragem verdadeiras.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Os dados brutos de sequenciamento para os quatro genomas antigos recentemente reconstruídos estão acessíveis no European Nucleotide Archive sob o número de acesso PRJEB62647 ( ERP147759 ). Informações detalhadas sobre a fonte do conjunto de dados de referência estão documentadas na Tabela Suplementar 3 . O alinhamento do genoma baseado em múltiplas referências, com e sem regiões de recombinação removidas, juntamente com árvores e arquivos de log para os resultados principais e todos os dados brutos e scripts necessários para reproduzir as análises para este estudo estão disponíveis em https://github.com/laduplessis /Pré-colombiano-Treponema-pallidum-do-Brasil ( https://doi.org/10.5281/zenodo.10063176 ).
Disponibilidade de código
Nenhum código interno especializado foi utilizado para este estudo. Todos os softwares utilizados para a análise dos dados neste estudo estão disponíveis publicamente e são citados no texto principal e nas informações suplementares. Os scripts customizados e o pipeline utilizado na análise e visualização da datação do relógio molecular estão depositados em https://github.com/laduplessis/Pre-Columbian-Treponema-pallidum-from-Brazil ( https://doi.org/10.5281/zenodo .10063176 ).
Referências
Crosby, AW O intercâmbio colombiano: consequências biológicas e culturais de 1492 (Greenwood Publishing Group, 2003).
Harper, KN, Zuckerman, MK, Harper, ML, Kingston, JD & Armelagos, GJ A origem e antiguidade da sífilis revisitadas: uma avaliação das evidências pré-colombianas do Velho Mundo para infecção treponêmica. Sou. J. Física. Antropol. 146 , 99–133 (2011).
Schuenemann, V. J. et al. Historic Treponema pallidum genomes from colonial Mexico retrieved from archaeological remains. PLoS Negl. Trop. Dis. 12, e0006447 (2018).
Majander, K. et al. Ancient bacterial genomes reveal a high diversity of Treponema pallidum strains in early modern Europe. Curr. Biol. 30, 3788–3803.e10 (2020).
Giffin, K. et al. A treponemal genome from an historic plague victim supports a recent emergence of yaws and its presence in 15th century Europe. Sci. Rep. 10, 9499 (2020).
Barquera, R. et al. Origem e estado de saúde dos africanos de primeira geração do México colonial. Curr. Biol. 30 , 2078–2091.e11 (2020).
Fenton, KA et al. Sífilis infecciosa em ambientes de alta renda no século XXI. Lanceta infecta. Dis. 8 , 244–253 (2008).
Beale, MA et al. A filogenia global das de Treponema pallidum linhagens revela a recente expansão e disseminação da sífilis contemporânea. Nat. Microbiol. 6 , 1549–1560 (2021).
Tsuboi, M. et al. Prevalência de sífilis entre homens que fazem sexo com homens: uma revisão sistemática global e meta-análise de 2000–20. Lanceta Globo. Saúde 9 , e1110–e1118 (2021).
Taouk, M. L. et al. Characterisation of Treponema pallidum lineages within the contemporary syphilis outbreak in Australia: a genomic epidemiological analysis. Lancet Microbe 3, e417–e426 (2022).
Tao, Y.-T. et al. Global, regional, and national trends of syphilis from 1990 to 2019: the 2019 global burden of disease study. BMC Public Health 23, 754 (2023).
Beale, MA et al. A epidemiologia genômica da sífilis revela o surgimento independente de resistência aos macrólidos em múltiplas linhagens circulantes. Nat. Comum. 10 , 3255 (2019).
Stamm, LV Sífilis: ressurgimento de um antigo inimigo. Micróbio. Fato celular. 3 , 363–370 (2016).
Mitja, O. et al. Re-emergência de bouba após tratamento com azitromicina em massa única seguido de tratamento direcionado: um estudo longitudinal. Lanceta 391 , 1599–1607 (2018).
Beale, MA et al. Reemergência de bouba e seleção de resistência bacteriana a medicamentos após administração em massa de azitromicina: uma investigação epidemiológica genômica. Lancet Microbe 1 , e263–e271 (2020).
Arora, N. et al. Origin of modern syphilis and emergence of a pandemic Treponema pallidum cluster. Nat. Microbiol. 2, 16245 (2016).
Vrbová, E. et al. Whole genome sequences of Treponema pallidum subsp. endemicum isolated from Cuban patients: the non-clonal character of isolates suggests a persistent human infection rather than a single outbreak. PLoS Negl. Trop. Dis. 16, e0009900 (2022).
Marks, M., Solomon, A. W. & Mabey, D. C. Endemic treponemal diseases. Trans. R. Soc. Trop. Med. Hyg. 109, 604 (2015).
Rothschild, B. M. History of syphilis. Clin. Infect. Dis. 40, 1454–1463 (2005).
Kanan, MW, Abbas, M. & Girgis, HY Bejel mutilante tardio nos beduínos nômades do Kuwait. Dermatologica 143 , 277-287 (1971).
Erdelyi, RL & Molla, AA Sífilis endêmica queimada (bejel). Plast. Reconst. Surg. 74 , 589–600 (1984).
Román, GC & Román, LN Ocorrência de complicações congênitas, cardiovasculares, viscerais, neurológicas e neuro-oftalmológicas na bouba tardia: um tema para pesquisas futuras. Rev. Infectar. Dis. 8 , 760–770 (1986).
Giacani, L. & Lukehart, SA As treponematoses endêmicas. Clin. Microbiol. Rev. 27 , 89–115 (2014).
Radolf, J. D. et al. Treponema pallidum, the syphilis spirochete: making a living as a stealth pathogen. Nat. Rev. Microbiol. 14, 744–759 (2016).
Baker, B. J. in The Routledge Handbook of Paleopathology (ed. Grauer, A. L.) Ch. 16 (2022); https://doi.org/10.4324/9781003130994-18.
Ortner, DJ Identificação de condições patológicas em restos de esqueletos humanos (Academic Press, 2003).
Forrestel, AK, Kovarik, CL & Katz, KA Sífilis sexualmente adquirida: aspectos históricos, microbiologia, epidemiologia e manifestações clínicas. Geléia. Acad. Dermatol. https://doi.org/10.1016/j.jaad.2019.02.073 (2020).
Harper, KN et al. Sobre a origem das treponematoses: uma abordagem filogenética. PLoS Negl. Tropa. Dis. 2 , e148 (2008).
de Melo, FL, de Mello, JCM, Fraga, AM, Nunes, K. & Eggers, S. Sífilis na encruzilhada da filogenética e da paleopatologia. PLoS Negl. Tropa. Dis. 4 , e575 (2010).
Filippini, J., Pezo-Lanfranco, L. & Eggers, S. Um estudo regional sistemático de treponematoses em sambaquis (sambaquis) brasileiros pré-colombianos. Chungara 51 , 403–425 (2019).
Meyer, M. & Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, db.prot5448 (2010).
Briggs, A. W. et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl Acad. Sci. USA 104, 14616–14621 (2007).
Adler, C. J., Haak, W., Donlon, D. & Cooper, A. Survival and recovery of DNA from ancient teeth and bones. J. Archaeol. Sci. 38, 956–964 (2011).
Dabney, J., Meyer, M. & Pääbo, S. Danos no DNA antigo. Primavera fria Harb. Perspectiva. Biol. 5 , a012567 (2013).
Akgul, G. et al. Inferir padrões de recombinação e divergência com genomas treponêmicos antigos e modernos. Pré-impressão em bioRxiv https://doi.org/10.1101/2023.02.08.526988 (2023).
Pla-Díaz, M. et al. Processos evolutivos no surgimento e disseminação recente do agente da sífilis, Treponema pallidum . Mol. Biol. Evol. 39 , msab318 (2022).
Strouhal, M. et al. Sequências completas do genoma de duas cepas de Treponema pallidum subsp. pertenue da Indonésia: estrutura modular de vários genes treponêmicos. PLoS Negl. Tropa. Dis. 12 , e0006867 (2018).
Mikalová, L. et al. Whole genome sequence of the Treponema pallidum subsp. endemicum strain Iraq B: a subpopulation of bejel treponemes contains full-length tprF and tprG genes similar to those present in T. p. subsp. pertenue strains. PLoS ONE 15, e0230926 (2020).
Powell, ML et al. O Mito da Sífilis: A História Natural da Treponematose na América do Norte (Univ. Press of Florida, 2005).
Baker, BJ et al. Avançar na compreensão da doença treponêmica no passado e no presente. Sou. J. Física. Antropol. 171 , 5–41 (2020).
Štaudová, B. et al. Sequência completa do genoma do Treponema pallidum subsp. cepa endemicum Bósnia A: o genoma está relacionado aos treponemas da bouba, mas contém poucos loci semelhantes aos treponemas da sífilis. PLoS Negl. Tropa. Dis. 8 , e3261 (2014).
Lieberman, NAP et al. O sequenciamento do genoma do Treponema pallidum de seis continentes revela variabilidade nos genes candidatos a vacina e dominância das cepas do clado Nichols em Madagascar. PLoS Negl. Tropa. Dis. 15 , e0010063 (2021).
Noda, AA et al. Bejel em Cuba: identificação molecular de Treponema pallidum subsp. endemicum em pacientes com diagnóstico de sífilis venérea. Clin. Microbiol. Infectar. 24 , 1210.e1–1210.e5 (2018).
Kawahata, T. et al. Bejel, a nonvenereal treponematosis, among men who have sex with men, Japan. Emerg. Infect. Dis. 25, 1581–1583 (2019).
Shinohara, K. et al. Clinical perspectives of Treponema pallidum subsp. endemicum infection in adults, particularly men who have sex with men in the Kansai area, Japan: a case series. J. Infect. Chemother. 28, 444–450 (2022).
Lewis, CM Jr, Akinyi, MY, DeWitte, SN & Stone, AC Patógenos antigos fornecem uma janela para a saúde e o bem-estar. Processo. Acad. Nacional. Ciência. EUA 120 , e2209476119 (2023).
Vågene, Å. J. et al. Genomas de Salmonella enterica de vítimas de uma grande epidemia do século XVI no México. Nat. Eco. Evol. 2 , 520–528 (2018).
Zhou, Z. et al. antiga e moderna A análise pan-genômica da Salmonella enterica demonstra a estabilidade genômica da linhagem invasora Para C por milênios. Curr. Biol. 28 , 2420–2428.e10 (2018).
Chave, FM et al. enterica adaptada ao homem O surgimento de Salmonella está ligado ao processo de neolitização. Nat. Eco. Evol. 4 , 324–333 (2020).
Haller, M. et al. Mass burial genomics reveals outbreak of enteric paratyphoid fever in the Late Medieval trade city Lübeck. iScience 24, 102419 (2021).
Schwarz, S., Skytte, L. & Rasmussen, K. L. Pre-Columbian treponemal infection in Denmark?—A paleopathological and archaeometric approach. Herit. Sci. 1, 19 (2013).
Rissech, C. et al. Esqueleto romano com possível treponematose no Nordeste da Península Ibérica: estudo morfológico e radiológico. Internacional J. Osteoarqueol. 23 , 651–663 (2013).
Gaul, JS, Grossschmidt, K., Gusenbauer, C. & Kanz, F. Um provável caso de sífilis congênita na Áustria pré-colombiana. Antropol. Anz. 72 , 451–472 (2015).
Raghavan, M. et al. Evidência genômica do Pleistoceno e da história populacional recente dos nativos americanos. Ciência 349 , aab3884 (2015).
Bardill, J. et al. Avançando a ética da paleogenômica. Ciência 360 , 384–385 (2018).
Blasis, P. A. D., Kneip, A. & Scheel-Ybert, R. Sambaquis e paisagem: dinâmica natural e arqueologia regional no litoral do sul do Brasil. Arqueología Suramericana 3 , 29–61 (2007).
Posth, C. et al. Reconstructing the deep population history of Central and South America. Cell 175, 1185–1197.e22 (2018).
Mitjà, O., Šmajs, D. & Bassat, Q. Advances in the diagnosis of endemic treponematoses: yaws, bejel, and pinta. PLoS Negl. Trop. Dis. 7, e2283 (2013).
Martins-Melo, FR, Ramos, AN Jr, Alencar, CH & Heukelbach, J. Mortalidade por doenças tropicais negligenciadas no Brasil, 2000–2011. Touro. Órgão Mundial da Saúde. 94 , 103–110 (2016).
Garra, KG et al. A escavação do Chaco Canyon revela preocupações éticas. Zumbir. Biol. 89 , 177–180 (2017).
Lima, T. A. Em busca dos frutos do mar os pescadores-coletores do litoral centro-sul do Brasil. Rev. USP https://doi.org/10.11606/issn.2316-9036.v0i44p270-327 (1999).
Gaspar, MD, DeBlasis, P., Fish, SK & Fish, PR em The Handbook of South American Archaeology (eds. Silverman, H. & Isbell, WH) 319–335 (Springer, 2008).
Fish, S. K., DeBlasis, P. & Gaspar, M. D. Eventos incrementais na construção de sambaquis, litoral sul do Estado de Santa Catarina. Rev. Mus. Arqueol. Etnol. https://doi.org/10.11606/issn.2448-1750.revmae.2000.109378 (2000).
Klokler, D. M. Food for Body and Soul: Mortuary Ritual in Shell Mounds (Laguna-Brazil). PhD thesis, Univ. of Arizona (2008).
Edwards, H. G. M. et al. Raman spectroscopic study of 3000-year-old human skeletal remains from a sambaqui, Santa Catarina, Brazil. J. Raman Spectrosc. 32, 17–22 (2001).
Beck, A. & Pereira, J. B. B. Variação do Conteúdo Cultural dos Sambaquis: Litoral de Santa Catarina. Doctoral thesis, Univ. of São Paulo (1972).
Prous, A. & Fogaça, E. Arqueologia da fronteira Pleistoceno-Holoceno no Brasil. Quat. Internacional 53-54 , 21–41 (1999).
Barbosa, P. N. A Coisa Ficou Preta: Estudo do Processo de Formação da Terra Preta do Sítio Arqueológico Jabuticabeira II. Doctoral thesis, Univ. of São Paulo (2007).
Okumura, MMM & Eggers, S. O povo da Jabuticabeira II: reconstrução do modo de vida em um sambaqui brasileiro. Homo 55 , 263–281 (2005).
Pezo-Lanfranco, L. et al. Desenvolvimento infantil, estresse fisiológico e expectativa de sobrevivência em pescadores-caçadores-coletores pré-históricos do sambaqui Jabuticabeira II, litoral sul do Brasil. PLoS ONE 15 , e0229684 (2020).
Rothschild, B. M. & Rothschild, C. Treponemal disease revisited: skeletal discriminators for yaws, bejel, and venereal syphilis. Clin. Infect. Dis. 20, 1402–1408 (1995).
Hajdas, I., Michczyński, A., Bonani, G., Wacker, L. & Furrer, H. Dating bones near the limit of the radiocarbon dating method: study case mammoth from Niederweningen, ZH Switzerland. Radiocarbon 51, 675–680 (2009).
Toso, A. et al. Intensificação da pesca como resposta à instabilidade socioecológica do Holoceno Superior no sudeste da América do Sul. Ciência. Rep. 11 , 23506 (2021).
Stuiver, M. & Reimer, PJ estendido 14 Banco de dados C e CALIB 3.0 revisado 14 Programa de calibração de idade C. Radiocarbono 35 , 215–230 (1993).
Heaton, TJ et al. Marine20 — A curva de calibração da idade do radiocarbono marinho (0–55.000 cal bp ). Radiocarbono 62 , 779–820 (2020).
Hogg, AG et al. Calibração SHCal20 Hemisfério Sul, 0–55.000 anos cal bp . Radiocarbono 62 , 759–778 (2020).
Reimer, PJ & Reimer, RW Um banco de dados de correção de reservatórios marinhos e interface on-line. Radiocarbono 43 , 461–463 (2001).
Ramsey, C. B. Bayesian analysis of radiocarbon dates. Radiocarbon 51, 337–360 (2009).
Pezo-Lanfranco, L., DeBlasis, P. & Eggers, S. Weaning process and subadult diets in a monumental Brazilian shellmound. J. Archaeol. Sci. 22, 452–469 (2018).
Dabney, J. & Meyer, M. in Methods in Molecular Biology, Vol. 1963 (eds Shapiro, B. et al.) 25–29 (Humana Press, 2019); https://doi.org/10.1007/978-1-4939-9176-1_4 (2019).
Kircher, M., Sawyer, S. & Meyer, M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 40, e3 (2012).
Wood, DE, Lu, J. & Langmead, B. Análise metagenômica aprimorada com Kraken 2. Genome Biol. 20 , 257 (2019).
Wingett, SW & Andrews, S. FastQ Screen: uma ferramenta para mapeamento multi-genoma e controle de qualidade. F1000Res 7 , 1338 (2018).
Kanz, C. et al. O banco de dados de sequências de nucleotídeos EMBL. Ácidos Nucleicos Res. 33 , D29–D33 (2005).
Sayers, EW et al. Recursos de banco de dados do Centro Nacional de Informações sobre Biotecnologia. Ácidos Nucleicos Res. 50 , D20–D26 (2022).
Schubert, M., Lindgreen, S. & Orlando, L. AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res. Notes 9, 88 (2016).
Valiente-Mullor, C. et al. One is not enough: on the effects of reference genome for the mapping and subsequent analyses of short-reads. PLoS Comput. Biol. 17, e1008678 (2021).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Neukamm, J., Peltzer, A. & Nieselt, K. DamageProfiler: fast damage pattern calculation for ancient DNA. Bioinformatics 37, 3652–3653 (2021).
Danecek, P. et al. Doze anos de SAMtools e BCFtools. Gigascience 10 , giab008 (2021).
Koboldt, DC et al. VarScan 2: descoberta de mutação somática e alteração do número de cópias em câncer por sequenciamento de exoma. Genoma Res. 22 , 568–576 (2012).
Quinlan, AR BEDTools: a ferramenta do Exército Suíço para análise de características do genoma. Curr. Protocolo. Bioinformática 47 , 11.12.1–11.12.34 (2014).
Milne, I. et al. Usando Tablet para exploração visual de dados de sequenciamento de segunda geração. Apresentação. Bioinformática 14 , 193–202 (2013).
Nakamura, T., Yamada, K. D., Tomii, K. & Katoh, K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 34, 2490–2492 (2018).
Larsson, A. AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30, 3276–3278 (2014).
Lechner, M. et al. Proteinortho: detection of (co-)orthologs in large-scale analysis. BMC Bioinformatics 12, 124 (2011).
Alikhan, N.-F., Petty, NK, Ben Zakour, NL & Beatson, SA BLAST Ring Image Generator (BRIG): comparações simples do genoma de procariontes. BMC Genômica 12 , 402 (2011).
Nguyen, L.-T., Schmidt, HA, von Haeseler, A. & Minh, BQ IQ-TREE: um algoritmo estocástico rápido e eficaz para estimar filogenias de máxima verossimilhança. Mol. Biol. Evol. 32 , 268–274 (2015).
Rodríguez, F., Oliver, JL, Marín, A. & Medina, JR O modelo estocástico geral de substituição de nucleotídeos. J. Teor. Biol. 142 , 485–501 (1990).
Estimativa filogenética de máxima verossimilhança a partir de sequências de DNA com taxas variáveis sobre locais: métodos aproximados. J. Mol. Evol. 39 , 306–314 (1994).
Gu, X., Fu, YX & Li, WH Estimativa de máxima verossimilhança da heterogeneidade da taxa de substituição entre sítios de nucleotídeos. Mol. Biol. Evol. 12 , 546–557 (1995).
Strimmer, K. & von Haeseler, A. Likelihood-mapping: a simple method to visualize phylogenetic content of a sequence alignment. Proc. Natl Acad. Sci. USA 94, 6815–6819 (1997).
Shimodaira, H. & Hasegawa, M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 16, 1114–1116 (1999).
Strimmer, K. & Rambaut, A. Inferring confidence sets of possibly misspecified gene trees. Proc. Biol. Sci. 269, 137–142 (2002).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Pinto, M. et al. não cultivável A análise em escala genômica do Treponema pallidum revela extensa variação genética dentro do paciente. Nat. Microbiol. 2 , 16190 (2016).
Grillová, L. et al. Genomas sequenciados diretamente de cepas contemporâneas de sífilis revelam diversidade impulsionada por recombinação em genes que codificam antígenos expostos à superfície previstos. Frente. Microbiol. 10 , 1691 (2019).
Addetia, A. et al. completa Estimativa da diversidade tprk em Treponema pallidum subsp. pálido . mBio 11 , e02726–20 (2020).
Liu, D. et al. Caracterização molecular baseada em esquemas de tipagem MLST e ECDC e análises de resistência a antibióticos de Treponema pallidum subsp. pallidum em Xiamen, China. Frente. Célula. Infectar. Microbiol. 10 , 618747 (2021).
Croucher, N. J. et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43, e15 (2015).
Didelot, X. & Wilson, D. J. ClonalFrameML: efficient inference of recombination in whole bacterial genomes. PLoS Comput. Biol. 11, e1004041 (2015).
Cejkova, D., Strouhal, M. & Smajs, D. Heterogeneidade genética intra-estirpe em Treponema pallidum ssp. pálido . Sexo. Transm. Infectar. 89 , A76 (2013).
Matějková, P. et al. Sequência completa do genoma de Treponema pallidum ssp. cepa SS14 pallidum determinada com matrizes de oligonucleotídeos. Microbiol BMC. 8 , 76 (2008).
Giacani, L. et al. Variação antigênica em Treponema pallidum : A diversidade da sequência TprK se acumula em resposta à pressão imunológica durante a sífilis experimental. J. Imunol. 184 , 3822–3829 (2010).
Smajs, D., Norris, SJ & Weinstock, GM Diversidade genética em Treponema pallidum : implicações para a patogênese, evolução e diagnóstico molecular de sífilis e bouba. Infectar. Geneta. Evol. 12 , 191–202 (2012).
de la Haba, R. R., Arahal, D. R., Márquez, M. C. & Ventosa, A. Phylogenetic relationships within the family Halomonadaceae based on comparative 23 S and 16 S rRNA gene sequence analysis. Int. J. Syst. Evol. Microbiol. 60, 737–748 (2010).
Yang, B., Wang, Y. & Qian, P.-Y. Sensitivity and correlation of hypervariable regions in 16 S rRNA genes in phylogenetic analysis. BMC Bioinformatics 17, 135 (2016).
Martijn, J. et al. Confident phylogenetic identification of uncultured prokaryotes through long read amplicon sequencing of the 16S-ITS-23S rRNA operon. Environ. Microbiol. 21, 2485–2498 (2019).
Bouckaert, R. et al. BEAST 2.5: Uma plataforma de software avançada para análise evolutiva Bayesiana. Computação PLoS. Biol. 15 , e1006650 (2019).
Drummond, AJ, Ho, SYW, Phillips, MJ & Rambaut, A. Filogenética relaxada e namoro com confiança. PLoS Biol. 4 , e88 (2006).
Drummond, AJ, Rambaut, A., Shapiro, B. & Pybus, OG Inferência coalescente bayesiana da dinâmica populacional passada a partir de sequências moleculares. Mol. Biol. Evol. 22 , 1185–1192 (2005).
Schuenemann, VJ et al. Genomas antigos revelam uma alta diversidade de Mycobacterium leprae na Europa medieval. PLoS Pathog. 14 , e1006997 (2018).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904 (2018).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer, 2016).
Yu, G., Smith, D. K., Zhu, H., Guan, Y. & Lam, T. T.-Y. ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8, 28–36 (2017).
Navascués, M., Depaulis, F. & Emerson, BC Combinando DNA contemporâneo e antigo em estudos genéticos e filogeográficos populacionais. Mol. Eco. Recurso. 10 , 760–772 (2010).
Duchêne, S., Duchêne, D., Holmes, EC & Ho, SYW O desempenho do teste de randomização de data em análises filogenéticas de dados de vírus estruturados no tempo. Mol. Biol. Evol. 32 , 1895–1906 (2015).
Rieux, A. & Balloux, F. Inferências de filogenias calibradas com ponta: uma revisão e um guia prático. Mol. Eco. 25 , 1911–1924 (2016).
Plummer, M., Martin, AD & Quinn, KM Aplicou inferência bayesiana em R usando MCMCpack. R Notícias 6 , 2–8 (2006).
Neukamm, J., Peltzer, A. & Nieselt, K. DamageProfiler.: fast damage pattern calculation for ancient DNA. Bioinformatics 37, 3652–3653 (2021).
Reconhecimentos
Os autores agradecem a I. Hajdas (ETH Zurique) pela realização do 14 C datação deste estudo e C. Steiner pelas ilustrações deste estudo; o Centro de Genômica Funcional de Zurique e o Biocentro de Viena para conduzir o sequenciamento de DNA; ao Museu de Arqueologia e Etnologia da Universidade de São Paulo pelo acesso ao material amostral do sítio Jabuticabeira II e, em especial, ao P. de Blasis pela aprovação da utilização das amostras ósseas, informações sobre a escavação original e aconselhamento sobre as considerações éticas considerando as conexões dos grupos indígenas modernos da região e do sítio arqueológico; e E. Krenak pelo aconselhamento profissional sobre como garantir a discrição para com os povos indígenas sul-americanos em questões de idioma e inclusão. Este trabalho foi apoiado pela Swiss National Science Foundation: concessão número 188963 “Rumo às origens da sífilis” (VJS e KM), o Programa Prioritário de Pesquisa Universitária da Universidade de Zurique 'Evolução em Ação: Dos Genomas aos Ecossistemas' (VJS), e pelos projetos BFU2017-89594R e PID2021-127010OB-100 do Ministério de Ciência e Inovação espanhol (FG-C. e MP-D.). MP-D. foi financiado pelo programa FPU17/02367 do Ministério de Educação espanhol. A LPL foi financiada pela FAPESP - 2017/17580-0.
Informação sobre o autor
Authors and Affiliations
Contribuições
V.J.S, F.G.-C. and K.M. conceived and led the investigation. V.J.S. and K.M. designed the study. S.E., L.P.L. and J.F. provided samples and archaeological contextualization. V.J.S. and F.G.-C. supervized the laboratory work, sampling or analysis and provided funding. K.M. and M.P.-D. performed laboratory experiments. M.P.-D., F.G.-C. and K.M. analysed the genetic data. L.d.P. and L.P.L. performed the molecular dating analysis. V.J.S., N.A., S.E. and J.F. aided with data interpretation. M.P-D. and L.dP. visualized the results. K.M., M.P.-D. and V.J.S. wrote the manuscript, with contributions from all co-authors.
Corresponding authors
Declarações éticas
Interesses competitivos
Os autores declaram não haver interesses conflitantes.
Revisão por pares
Informações de revisão por pares
A Nature agradece a Maarten Blaauw, James McInerney, Hendrik Poinar e ao(s) outro(s) revisor(es) anônimo(s) por sua contribuição para a revisão por pares deste trabalho.
Informações adicionais
Nota do editor A Springer Nature permanece neutra em relação a reivindicações jurisdicionais em mapas publicados e afiliações institucionais.
Figuras e tabelas de dados estendidas
Dados estendidos Fig. 1 Curvas de datação 14C calibradas de amostras usadas para reconstrução de genoma antigo e perfis de danos para autenticação de aDNA.
Calibration conducted by Calib radiocarbon calibration program, showing the a) pre- and b) post-reservoir effect corrected (modelled) curves for the four samples (ZH1390, ZH1540, ZH141 and ZH1557) included in the genomic analyses. Damage profiles obtained with the DamageProfiler tool130 show the misincorporation patterns induced by age, for each sample that yielded a reconstructed genome: c) ZH1540, d) ZH1390, e) ZH1541 and f) ZH1557. A pattern of cytosine-to-thymine base misincorporation accumulating at the end of the reads is indicative of authentic ancient DNA in the sample.
Extended Data Fig. 2 Comparison of the topologies of maximum-likelihood (ML) trees.
a) ML tree topology with all genes included in the multiple genome alignment using GTR + G + I as the evolutionary model and 1000 bootstrap repetitions. b) ML tree topology after excluding tp0897, tp0316 and tp0317 and recombinant genes from the multiple genome alignment using GTR + G + I as the evolutionary model and 1000 bootstrap repetitions. The different clades corresponding to TPE and TEN, and the Nichols and SS14 lineages of TPA are colour-coded according to the legend. Bootstrap support values higher than 70% are indicated by pink circles, with circle size proportional to bootstrap support. The subclade SS14-Ω, that includes all SS14-lineage TPA strains except ancient TPA strains, MD06B, MD18Be and MexicoA, is collapsed.
Dados estendidos Fig. 3 Árvore de máxima verossimilhança (ML) com genes recombinantes, hipervariáveis e 16S, 23S excluídos.
Árvore ML obtida após exclusão de tp0897 , tp0316, tp0317, 16 S, 23 S e genes recombinantes do alinhamento múltiplo do genoma usando GTR + G + I como modelo evolutivo e 1000 repetições de bootstrap. Os diferentes clados correspondentes a TPE e TEN, e as linhagens Nichols e SS14 de TPA são codificados por cores de acordo com a legenda. Valores de suporte de bootstrap superiores a 70% são indicados por círculos rosa, com tamanho de círculo proporcional ao suporte de bootstrap.
Dados Estendidos Fig. 4 Divergência raiz-a-ponta em relação à data de amostragem de cepas de T. pallidum e respectivos coeficientes de correlação de Pearson.
As áreas sombreadas indicam o intervalo de confiança de 95% das linhas de regressão. a ) Conjunto de dados completo. b ) Apenas cepas modernas. c ) Apenas cepas clínicas modernas. d ) Apenas cepas modernas de TPE. e ) Apenas cepas modernas de RTE. f ) Apenas cepas modernas de TPA SS14. g ) Apenas cepas modernas de TPA Nichols. h ) Apenas cepas clínicas modernas de TPA Nichols.
Extended Data Fig. 5 Posterior densities of the times of the most recent ancestors (tMRCAs) of the T. pallidum subspecies and major lineages as estimated by the molecular clock dating under different relaxed clock models and the results of the Bayesian date randomization test (DRT).
a) TPE, b) TPA, c) TPA SS14, d) TPA Nichols, e) TEN and f) T. pallidum. Vertical lines inside the density curves in panels a-f indicate the upper and lower limits of the 95% HPD intervals. g) Posterior distributions for the mean clock rate, using the true, unpermuted sampling dates (far left) and 50 replicates with sampling dates permuted among tips under a UCED model with a narrow mean clock-rate prior. Distributions are truncated at the upper and lower limits of the 95% HPD intervals and horizontal red lines indicate the median estimates. The red dashed lines indicate the median and the limits of the 95% HPD interval of the mean clock rate estimated under the true sampling dates.
Dados estendidos Fig. 6 Árvore de credibilidade máxima do clado (MCC) da análise de datação do relógio molecular do conjunto de dados de contexto do genoma 98 com ZH1540 incluído (n = 99).
A inserção mostra uma visão simplificada de toda a árvore, com a linha tracejada indicando a parte da árvore mostrada na figura principal. Genomas antigos são rotulados em negrito e ZH1540 é marcado por uma estrela. As barras azuis indicam os intervalos HPD de 95% das idades dos nós e o texto em vermelho a probabilidade posterior de um clado ser monofilético (mostrado apenas para nós com probabilidade posterior> 0,8).
Dados estendidos Fig. 7 Árvore de credibilidade máxima do clado (MCC) da análise de datação do relógio molecular do conjunto de dados de contexto do genoma 98 com ZH1540 excluído (n = 98).
A inserção mostra uma visão simplificada de toda a árvore, com a linha tracejada indicando a parte da árvore mostrada na figura principal. Genomas antigos são rotulados em negrito. As barras azuis indicam os intervalos HPD de 95% das idades dos nós e o texto em vermelho a probabilidade posterior de um clado ser monofilético (mostrado apenas para nós com probabilidade posterior> 0,8).
Nenhum comentário:
Postar um comentário
Observação: somente um membro deste blog pode postar um comentário.