História genética de um grupo arcaico de hominídeos da Caverna Denisova na Sibéria
Nature volume 468 , páginas 1053–1060 ( 2010 )
Resumo
Utilizando DNA extraído de um osso de dedo encontrado na Caverna Denisova, no sul da Sibéria, sequenciamos o genoma de um hominídeo arcaico com uma cobertura de cerca de 1,9 vezes. Este indivíduo pertence a um grupo que compartilha uma origem comum com os neandertais. Esta população não esteve envolvida no suposto fluxo gênico dos neandertais para os eurasianos; no entanto, os dados sugerem que ela contribuiu com 4 a 6% de seu material genético para os genomas dos melanésios atuais. Designamos esta população de hominídeos como "denisovanos" e sugerimos que ela pode ter sido disseminada na Ásia durante o Pleistoceno Superior. Um dente encontrado na Caverna Denisova carrega um genoma mitocondrial muito semelhante ao do osso do dedo. Este dente não compartilha características morfológicas derivadas com os neandertais ou os humanos modernos, indicando ainda mais que os denisovanos têm uma história evolutiva distinta dos neandertais e dos humanos modernos.
Conteúdo semelhante sendo visualizado por outras pessoas


Principal
Há menos de 200.000 anos, humanos anatomicamente modernos (ou seja, humanos com esqueletos semelhantes aos dos humanos atuais) surgiram na África. Naquela época, bem como posteriormente, quando os humanos modernos surgiram na Eurásia, outros hominídeos "arcaicos" já estavam presentes na Eurásia. Na Europa e na Ásia Ocidental, os hominídeos definidos como neandertais com base em sua morfologia esquelética viveram há pelo menos 230.000 anos, antes de desaparecerem do registro fóssil há cerca de 30.000 anos. 1 No leste da Ásia, não há consenso sobre quais grupos estavam presentes. Por exemplo, na China, alguns enfatizaram afinidades morfológicas entre os neandertais e o espécime de Maba. 2 , ou entre o Homo heidelbergensis e o crânio de Dali 3 . No entanto, outros classificam esses espécimes como ' Homo sapiens primitivos ' 4 . Além disso, até pelo menos 17.000 anos atrás, o Homo floresiensis , um hominídeo de baixa estatura que parece representar uma divergência inicial da linhagem que levou aos humanos atuais 5 , 6 , 7 , estava presente na ilha de Flores, na Indonésia, e possivelmente em outros lugares.
Sequências de DNA recuperadas de restos mortais de hominídeos oferecem uma abordagem complementar à morfologia para a compreensão das relações entre hominídeos. Para os neandertais, o genoma nuclear foi recentemente determinado com uma cobertura de cerca de 1,3 vezes maior. 8 Isso revelou que as sequências de DNA neandertais e as dos humanos atuais compartilham ancestrais comuns, em média, há cerca de 800.000 anos, e que a divisão populacional dos ancestrais neandertais e humanos modernos ocorreu entre 270.000 e 440.000 anos atrás. Também demonstrou que os neandertais compartilhavam mais variantes genéticas com os humanos atuais na Eurásia do que com os humanos atuais na África Subsaariana, indicando que o fluxo gênico dos neandertais para os ancestrais de não africanos ocorreu a tal ponto que 1% a 4% dos genomas de pessoas fora da África são derivados de neandertais. 8 . Além disso, dez sequências parciais e seis completas de DNA mitocondrial (mt) foram determinadas em neandertais 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 . Isso mostrou que todos os neandertais estudados até agora compartilham um ancestral comum de mtDNA da ordem de 100.000 anos atrás 10 , e por sua vez, compartilham um ancestral comum com os mtDNAs dos humanos atuais, há cerca de 500.000 anos 10 , 18 , 19 (como esperado, isso é mais antigo do que o tempo de divisão da população humana moderna entre Neandertais de 270.000 e 440.000 anos atrás, estimado a partir do genoma nuclear 8 ). Uma dessas sequências de mtDNA também mostrou que hominídeos portadores de mtDNAs típicos de neandertais estavam presentes tão a leste quanto as montanhas Altai, no sul da Sibéria. 13 .
Em 2008, a falange manual distal de um hominídeo juvenil foi escavada na Caverna Denisova. Este sítio arqueológico está localizado nas Montanhas Altai, no sul da Sibéria, e é um sítio de referência para o Paleolítico Médio e Superior da região, onde escavações sistemáticas ao longo dos últimos 25 anos revelaram camadas culturais que indicam que a ocupação humana no local começou há cerca de 280.000 anos. 20 A falange foi encontrada na camada 11, datada de 50.000 a 30.000 anos atrás. Esta camada contém microlâminas e ornamentos corporais de pedra polida, típicos da "indústria do Paleolítico Superior", geralmente considerada associada aos humanos modernos, mas também ferramentas de pedra mais características do Paleolítico Médio inicial, como raspadores laterais e blocos de Levallois. 21 , 22 , 23 .
Recentemente, usamos uma abordagem de captura de DNA 10 em combinação com sequenciamento de alto rendimento para determinar um genoma completo de mtDNA da falange de Denisova. Surpreendentemente, este mtDNA divergiu da linhagem comum que levou aos mtDNAs humanos e neandertais modernos há cerca de um milhão de anos. 19 , ou seja, cerca de duas vezes mais distante no tempo do que a divergência entre os mtDNAs de Neandertais e humanos modernos.
No entanto, o mtDNA é herdado maternalmente como uma unidade única sem recombinação e, portanto, está sujeito a eventos aleatórios, como deriva genética, bem como fluxo gênico e seleção positiva. Em contraste, o genoma nuclear compreende dezenas de milhares de loci não ligados, em sua maioria de evolução neutra. Isso permite análises de relações genéticas que são robustas à estocasticidade da deriva genética e são muito menos afetadas pela seleção positiva. Para esclarecer a relação do indivíduo Denisova com outros grupos de hominídeos, sequenciamos o genoma nuclear de Denisova e analisamos suas relações genômicas com os neandertais e os humanos atuais. Também tentamos esclarecer a cronologia da ocupação hominídea da caverna e identificamos um dente desse grupo de hominídeos entre o material escavado na Caverna Denisova.
Determinação da sequência de DNA
Toda a porção interna da amostra da falange foi usada para extração de DNA em nossa instalação de sala limpa, onde procedimentos para minimizar a contaminação do DNA humano atual são rigorosamente implementados. 24 , 25 ( Seção 1 de Informações Suplementares ). O DNA foi tratado com duas enzimas: uracil-DNA-glicosilase, que remove resíduos de uracila do DNA para deixar sítios abásicos 26 e a endonuclease VIII, que corta o DNA nos lados 5' e 3' dos sítios abásicos. A incubação subsequente com a polinucleotídeo quinase T4 e a DNA polimerase T4 foi utilizada para gerar extremidades rombas fosforiladas em 5', passíveis de ligação com adaptadores. Como a grande maioria dos resíduos de uracila ocorre próximo às extremidades de moléculas de DNA antigas, esse procedimento leva a uma redução apenas moderada no comprimento médio das moléculas na biblioteca, mas a uma redução significativa na incorporação incorreta de nucleotídeos derivados de uracila. 27 .
Duas bibliotecas de sequenciamento independentes (SL3003 e SL3004) foram criadas a partir do DNA, usando um protocolo Illumina modificado 28 onde uma reação em cadeia da polimerase (PCR) é usada para adicionar um índice de 7 nucleotídeos (neste caso, 5'-GTCGACT-3') às moléculas da biblioteca. Este índice garante que as bibliotecas não sejam contaminadas por outras bibliotecas de sequenciamento quando retiradas da sala limpa para serem sequenciadas. 29
As bibliotecas foram sequenciadas na plataforma Illumina Genome Analyser IIx por 101 ciclos a partir de cada extremidade das moléculas e mais 7 ciclos para determinação do índice até que quase todas as sequências únicas nas bibliotecas tivessem sido observadas múltiplas vezes, ou seja, quase todos os clones presentes nas bibliotecas tivessem sido sequenciados ( seção 1 de Informações Suplementares ). As bases foram chamadas utilizando o algoritmo de aprendizado de máquina Ibis. 30 e uma sobreposição de pelo menos 11 bases foi necessária para que as leituras de extremidade pareada fossem fundidas a sequências de DNA de tamanho molecular completo que foram analisadas posteriormente. Isso resulta em uma taxa de erro bastante reduzida. 27 , embora remova da análise as poucas moléculas com mais de 191 nucleotídeos de comprimento ( ∼ 0,1% em SL3003 e ∼ 0,2% em SL3004). As sequências foram mapeadas usando o programa BWA. 31 aos genomas humano (hg18/NCBI 36) e do chimpanzé (panTro2/CGSC 2.1), bem como ao genoma ancestral inferido dessas espécies (do alinhamento de seis vias Enredo-Pecan-Ortheus) 32 . Duplicatas de PCR foram identificadas e usadas para aumentar ainda mais a precisão da sequência, chamando sequências de consenso.
Um total de 82.227.320 sequências mapeadas exclusivamente (qualidade de mapeamento ≥ 30) para o genoma humano, resultando em cerca de 5,2 gigabases de sequências de DNA (cobertura genômica 1,9 vezes maior), e 72.304.848 sequências mapeadas exclusivamente para o genoma do chimpanzé. Quando as substituições inferidas como tendo ocorrido nas linhagens Denisova e humana atual foram comparadas, os números relativos de diferentes classes de substituições de nucleotídeos são notavelmente semelhantes, e o excesso de substituições candidatas na linhagem Denisova em relação à linhagem humana atual é de apenas 1,7 vezes ( Figura 2.2 e Tabela 2.4 Complementares ). Isso reflete uma melhora na taxa de erro em relação ao genoma neandertal em mais de uma ordem de magnitude. 8 e é principalmente devido à remoção enzimática de resíduos de uracila do DNA de Denisova 27 . Estimamos que a maioria dos erros nas sequências de DNA de Denisova se deve à baixa cobertura genômica e não a quaisquer características típicas do DNA antigo.
Estimativas de contaminação por DNA humano
Embora medidas rigorosas para prevenir a contaminação dos experimentos com DNA de humanos atuais tenham sido implementadas em todas as etapas do laboratório, é impossível prevenir completamente a contaminação, pois amostras de ossos e reagentes podem ser contaminados antes de entrarem na sala limpa. Para estimar os níveis de contaminação nas sequências produzidas, utilizamos três abordagens ( Informações Suplementares, seção 3).
Primeiramente, estimamos o nível de contaminação por mtDNA usando 276 posições de sequência onde o mtDNA de Denisova difere de >99% dos mtDNAs humanos atuais. Para a biblioteca SL3003, observamos 7.433 sequências únicas que cobriam tais posições, e 7.421 eram do tipo Denisova. Para a biblioteca SL3004, os números correspondentes foram 5.042 e 5.036, indicando que a contaminação por mtDNA nas bibliotecas é da ordem de 0,2% (intervalo de confiança de 95% [IC]: 0,1-0,3%) e 0,1% (IC: 0,1-0,3%), respectivamente.
Em segundo lugar, identificámos sequências que são exclusivas do cromossoma Y 8 Se o indivíduo de quem a falange deriva for do sexo feminino, o número dessas sequências representa a extensão da contaminação por DNA masculino. Encontramos zero e três dessas sequências do cromossomo Y nas duas bibliotecas, respectivamente, enquanto 1.449 e 696 são esperadas se o indivíduo for do sexo masculino. Assim, a contaminação por DNA do osso derivado de uma falange feminina e masculina nas duas bibliotecas é da ordem de 0,00% (IC: 0,00-0,25%) e 0,43% (IC: 0,09-1,26%), respectivamente.
Terceiro, para estimar a extensão da contaminação nuclear do DNA, usamos uma biblioteca para identificar posições onde o indivíduo Denisova carrega uma variante de sequência ancestral, isto é, semelhante à dos chimpanzés, que entre os humanos atuais é derivada e não se sabe que varia. Em seguida, examinamos sequências que mapeiam nessas posições na outra biblioteca e determinamos se elas carregam a sequência ancestral ou a sequência derivada. A observação de uma sequência derivada na segunda biblioteca pode ser devido a uma de três possibilidades: que o fragmento de DNA em questão vem da contaminação humana atual; que o indivíduo Denisova é heterozigoto na posição em questão; ou que houve um erro de sequenciamento. Implementamos um método de máxima verossimilhança que usa o número de observações independentes de estados ancestrais e derivados entre posições para coestimar a contaminação juntamente com a heterozigosidade e o erro de sequenciamento como parâmetros incômodos ( seção 3 de Informações Suplementares ). A partir dessa análise, infere-se que ambas as bibliotecas têm taxas de contaminação inferiores a 1%.
Características ancestrais e duplicações
O rascunho da sequência do genoma Denisova permite a identificação de características ancestrais no genoma Denisova e derivadas de humanos atuais. Descrevemos anteriormente um conjunto de 10,5 milhões de diferenças em nucleotídeos únicos e cerca de meio milhão de inserções/deleções (indels) inferidas como decorrentes de mudanças ocorridas na linhagem humana desde a separação do ancestral comum com o chimpanzé. 8 . Destas, 4.267.431 (40,5%) diferenças de nucleotídeo único e 105.372 (22,0%) indels são cobertas pelas sequências Denisova. Identificamos 129 substituições de aminoácidos inferidas e 14 indels nas sequências codificadoras de genes onde o indivíduo Denisova carrega os alelos ancestrais em posições onde os humanos atuais carregam alelos derivados e não são conhecidos por variar ( Seção 4 de Informações Suplementares ). Também identificamos 90 desses locais em regiões não traduzidas (UTRs) 5', 392 em UTRs 3', dois em genes de microRNA e 104 em regiões aceleradas humanas. Quando comparamos os genomas Denisova e Neandertal, descobrimos que eles carregam o mesmo estado atribuído em diferenças de nucleotídeo único em 87,9% das posições ancestrais e 97,7% das posições derivadas. Os resultados para indels são semelhantes: 87,6% para estados ancestrais e 98,6% para estados derivados ( Tabela Suplementar 4.3 ).
Analisamos o conteúdo de duplicação segmentar do genoma Denisova detectando regiões com excesso de profundidade de leitura ( seção 5 de Informações Suplementares ). Em uma comparação tripla dos genomas Denisova, Neandertal e humano atual, encontramos um excesso de duplicações privadas de Denisova (2,27 megabases (Mb)) em comparação com duplicações que eram privadas em Neandertais (0,60 Mb) ou humanos atuais (1,32 Mb). Essas regiões foram identificadas com base em assinaturas de excesso de profundidade de leitura e divergência de sequência aumentada, tornando improvável que sejam artefatos. Também identificamos duas regiões onde a arquitetura de duplicação de Denisova é mais semelhante à do chimpanzé do que à dos Neandertais ou humanos atuais, incluindo duas regiões cromossômicas associadas à doença neurológica em humanos: atrofia muscular espinhal em 5q13 (incluindo SMN2 , uma das duplicações genéticas mais recentes na linhagem humana) e doença neuropsiquiátrica em 16p12.1.
Relationship to Neanderthals and modern humans
Uma questão fundamental é se o indivíduo Denisova é um grupo externo aos neandertais e aos humanos modernos, como sugere o mtDNA 19 , seja um grupo irmão dos neandertais ou dos humanos modernos, ou se se enquadra na faixa de variação de qualquer um desses dois grupos. Abordamos isso estimando a divergência entre a sequência de referência do genoma Denisova e do humano como uma fração da divergência entre os humanos atuais e o ancestral comum compartilhado com o chimpanzé. Para isso, pontuamos a frequência com que o genoma Denisova carrega o estado humano versus o estado do chimpanzé em posições onde os genomas de referência humano e do chimpanzé diferem; assumindo taxas evolutivas constantes ( seção 2 de Informações Suplementares ). Restringimos esta análise às partes do genoma de referência humano que são de ancestralidade africana. 33 como fluxo genético dos neandertais para os não africanos 8 poderia complicar essas análises. O genoma de Denisova divergiu do genoma humano de referência em 11,7% (IC: 11,4-12,0%) ao longo da linhagem até o ancestral humano-chimpanzé. Para o Neandertal Vindija, a divergência é de 12,2% (IC: 11,9-12,5%). Assim, enquanto a divergência do mtDNA de Denisova com os mtDNAs humanos atuais é cerca de duas vezes mais profunda que a do mtDNA Neandertal 19 , a divergência média do genoma nuclear de Denisova dos humanos atuais é semelhante à dos neandertais.
Uma possível explicação para a divergência semelhante entre o indivíduo Denisova e os neandertais dos africanos atuais é que ambos descendem de uma população ancestral comum que se separou anteriormente dos ancestrais dos humanos atuais. Tal cenário preveria uma relação mais próxima entre o indivíduo Denisova e os neandertais do que entre qualquer um deles e os humanos atuais. Para testar essa previsão, estimamos a divergência entre pares de sete genomas antigos e modernos (Denisova, Neandertais, Franceses, Han, Papuas, Iorubás e San), usando uma abordagem em que corrigimos as taxas de erro em cada genoma com base na suposição de que cada um tem o mesmo número de diferenças verdadeiras em relação ao chimpanzé ( seção 6 de Informações Suplementares ). A divergência média entre os neandertais Denisova e Vindija é estimada em 9,84% do caminho até o ancestral chimpanzé-humano; ou seja, menos do que a divergência média de 12,38% de ambos em relação aos africanos atuais. Considerando 6,5 milhões de anos para a divergência entre humanos e chimpanzés, isso implica que as sequências de DNA dos neandertais e do indivíduo denisovano divergiram em média há 640.000 anos, e dos africanos atuais há 804.000 anos.
Para analisar mais a fundo a relação do indivíduo Denisova e dos neandertais, alinhamos as sequências de Denisova, Neandertal e Iorubá ao genoma do chimpanzé, escolhemos uma única sequência aleatoriamente para representar cada grupo e examinamos locais onde foram observadas duas cópias de um alelo derivado e uma cópia de um alelo ancestral. Espera-se que erros de sequenciamento tenham uma contribuição insignificante nesses locais. O número de locais onde o indivíduo Denisova e o Neandertal se agrupam, excluindo os Iorubás e os chimpanzés, é de 46.362, em comparação com uma média de 22.012 locais para os outros dois padrões possíveis (Iorubá e Denisova, ou Iorubá e Neandertal). Esse excesso de locais onde Denisova e Neandertal se agrupam apoia a visão de que o indivíduo Denisova e os neandertais compartilham uma história comum desde a separação dos ancestrais dos humanos modernos ( seção 6 de Informações Suplementares ).
Um gargalo específico do Neandertal
O fato de que o genoma nuclear de Denisova compartilha, em média, um ancestral comum mais recente com os neandertais do que com os humanos atuais levanta a questão de se a divergência geral da sequência de DNA do indivíduo de Denisova se enquadra dentro do grupo morfológica e geograficamente definido como neandertais, ou se representa um grupo irmão dos neandertais.
Para investigar esta questão, aproveitámos o facto de que, além dos três indivíduos da Caverna de Vindija, na Croácia, de onde foram produzidas a maior parte das sequências do genoma neandertal, determinámos sequências de ADN nuclear de mais três indivíduos neandertais da Rússia, Espanha e Alemanha. 8 Destes, o esqueleto de uma criança neandertal, com 60.000 a 70.000 anos, encontrado na Caverna Mezmaiskaya, na Rússia, é o mais antigo e geograficamente mais próximo do indivíduo de Denisova. Utilizando os 56 Mb de sequências de DNA autossômico determinadas a partir deste espécime 8 , estimamos que a divergência na sequência de DNA entre os neandertais de Vindija e Mezmaiskaya corresponde a uma data de 140.000 ± 33.000 anos atrás ( seção 6 de Informações Suplementares ) ( Fig. 1 ). Essa divergência notavelmente baixa — que corresponde a cerca de um terço do par mais próximo de humanos atuais que analisamos — está de acordo com a observação de que a diversidade entre os mtDNAs neandertais é baixa em relação aos humanos atuais. 10 e indica que os neandertais Vindija e Mezmaiskaya descendem de uma população ancestral comum que passou por um gargalo drástico desde que se separou dos ancestrais do indivíduo Denisova.
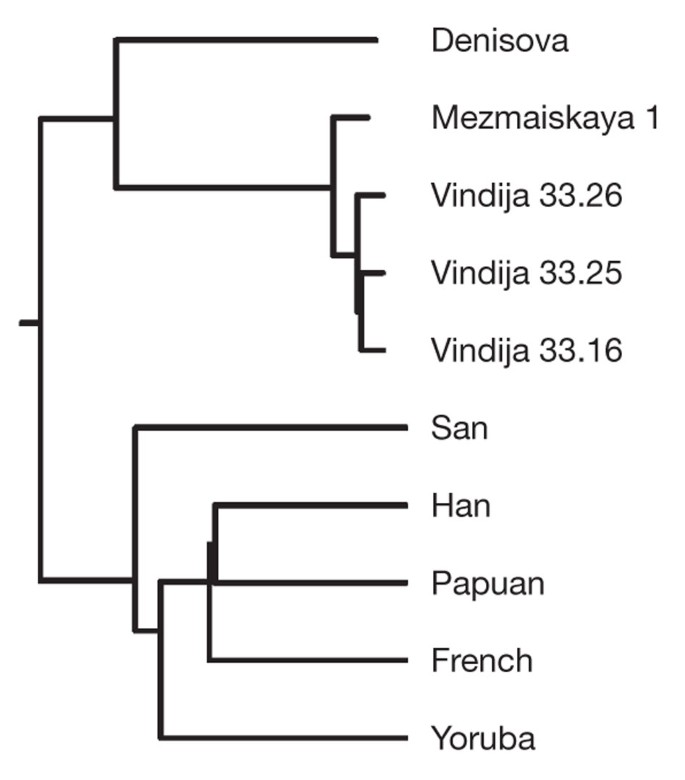
Vindija 33.16, Vindija 33.25 e Vindija 33.26 referem-se aos números de catálogo dos ossos de neandertal.
Para compreender melhor o gargalo na história dos neandertais de Vindija e Mezmaiskaya, examinamos alinhamentos quádruplos da sequência genômica do neandertal de Vindija, do neandertal de Mezmaiskaya, do indivíduo de Denisova e do genoma do chimpanzé. Em substituições transversais, onde duas cópias dos alelos derivados são observadas, detectamos 924 substituições que agrupam os neandertais de Vindija e Mezmaiskaya, 80 que agrupam Vindija e Denisova e 81 que agrupam Mezmaiskaya e Denisova. Isso corresponde a pelo menos 65% de probabilidade de que as sequências de DNA nos neandertais compartilhem um ancestral comum mais recente do que sua separação do ancestral do indivíduo de Denisova ( Informações Suplementares , seção 7). É muito maior do que a probabilidade de 15 a 20% associada ao gargalo "Fora da África", comum aos não africanos atuais. 34 . Se substituirmos o Neandertal de Mezmaiskaya nesta análise por um Neandertal de El Sidron, Espanha, ou de Feldhofer, Alemanha, os resultados são qualitativamente semelhantes, embora os números sejam menores ( seção 7 de Informações Suplementares ). Assim, concluímos que os Neandertais tardios em uma ampla distribuição geográfica têm uma história populacional distinta daquela do indivíduo Denisova, pois compartilham um forte gargalo populacional não experimentado pelos ancestrais do indivíduo Denisova. Chamamos o grupo ao qual esse indivíduo pertencia de Denisovanos em analogia aos Neandertais, pois os Denisovanos são descritos pela primeira vez com base em dados moleculares da Caverna Denisova, assim como os Neandertais foram descritos pela primeira vez com base em restos de esqueletos recuperados no Vale do Neander, na Alemanha.
Nenhum fluxo genético denisovano em todos os eurasianos
Mostramos anteriormente que os neandertais de Vindija compartilham mais alelos derivados com não africanos do que com africanos, o que é consistente com os neandertais contribuindo com 1–4% dos genomas dos humanos atuais na Eurásia. 8. To investigate the extent to which the Denisova individual shares this pattern, we examined alignments of sets of four genomes, each consisting of an African (Yoruba or San), a Eurasian (French or Han), an archaic hominin (Neanderthal or Denisovan) and the chimpanzee. We randomly sampled one allele from each of the three hominins, and counted all transversion differences between the African and the Eurasian where the archaic individual carries the derived allele (the ‘D statistics’ of ref. 8). Neanderthals match the French genome on average 4.6 ± 0.7% more often than they match the Yoruba genome (Table 1). Although the Denisova individual also matches the French more than the Yoruba genome, this skew is significantly less strong at 1.8 ± 0.5%. The estimates of D statistics were quantitatively consistent (within two standard deviations) for all other choices of Eurasian and African populations (Table 1). These findings indicate that the archaic component of the Eurasian gene pool is less closely related to the Denisova individual than to Neanderthals.
Também examinamos 13 regiões genômicas que identificamos anteriormente como candidatas a uma contribuição de material genético arcaico para os não africanos, com base em suas divergências genéticas mais profundas nos não africanos do que nos africanos. 8 Utilizando "SNPs de marcação" que informam se um haplótipo é da linhagem exclusiva de não africanos, descobrimos que o indivíduo Denisova corresponde ao haplótipo não africano profundamente divergente em 6 casos, enquanto os Neandertais o fazem em 11 casos ( Informações Suplementares , seção 7). Assim, tanto os Neandertais quanto os Denisovanos estão mais relacionados do que seria de se esperar por acaso a esses segmentos genômicos, mas o sinal nos Denisovanos é mais fraco.
Essas análises indicam que os neandertais são mais intimamente relacionados do que os denisovanos à população que contribuiu para o pool genético dos ancestrais dos eurasianos atuais. O fato de os eurasianos compartilharem alguma afinidade adicional com o indivíduo denisovano em relação aos africanos é compatível com um cenário em que os denisovanos compartilhavam parte de sua história com os neandertais antes do fluxo gênico dos neandertais para os humanos modernos.
Fluxo genético denisovano nos ancestrais dos melanésios
Embora o indivíduo Denisova derive de uma população que não esteve diretamente envolvida no fluxo gênico dos neandertais para os eurasianos, é possível que os denisovanos tenham se misturado com os ancestrais dos povos atuais em algumas partes do Velho Mundo. Para investigar isso, analisamos a relação do genoma Denisova com os genomas de 938 humanos atuais de 53 populações que foram genotipadas em 642.690 polimorfismos de nucleotídeo único (SNPs). 35 . Classificamos cada um desses humanos atuais com base em sua proximidade relativa aos neandertais e ao indivíduo de Denisova em posições onde temos dados de alta qualidade para os genomas neandertais e de Denisova ( seção 8 de Informações Suplementares ). Usando as médias das 53 populações, os dois primeiros componentes principais separam as populações em três grupos ( Fig. 2 ): primeiro, as 7 populações da África Subsaariana; segundo, um grupo de 44 populações não africanas, bem como um grupo do norte da África; e terceiro, populações de Papua e Bougainville da Melanésia. Quando indivíduos de populações selecionadas são analisados separadamente, os ilhéus de Papua e Bougainville permanecem distintos de quase todos os indivíduos fora da África ( Fig. 8.1b Suplementar ). Assim, com relação à sua relação com os neandertais e os denisovanos, as populações da Melanésia se destacam em relação a outras populações não africanas.

Análise de componentes principais das médias de 53 populações humanas atuais projetadas sobre os dois principais componentes definidos por Denisova: neandertais e chimpanzés. As sete populações "africanas" são San, Mbuti, Biaka, Bantu do Quênia, Bantu da África do Sul, Iorubá e Mandenka; as populações "não africanas" são 44 grupos diversos de fora da África, exceto os papuas e os ilhéus de Bougainville.
Para explorar isso mais a fundo, analisamos a relação do genoma de Denisova com os genomas de cinco humanos atuais que sequenciamos anteriormente para uma cobertura cerca de cinco vezes maior. 8 (um genoma Yoruba e um San da África, um genoma Francês da Europa, um genoma Han da China e um genoma Papua da Melanésia), bem como sete humanos atuais que sequenciamos para uma cobertura de 1 a 2 vezes para este estudo (um genoma Mbuti da África, um genoma Sardenho da Europa, um genoma Mongol da Ásia Central, um genoma Cambojano do Sudeste Asiático, um genoma Papua adicional da Melanésia, um genoma de um ilhéu de Bougainville da Melanésia e um genoma Karitiana da América do Sul) ( seção 9 de Informações Suplementares ). Usamos a D estatística 8 para testar se vários pares de humanos atuais compartilham números iguais de alelos derivados com o indivíduo Denisova. Para fazer isso, restringimos as comparações a pares de humanos atuais sequenciados ao mesmo tempo para minimizar a chance de que diferenças no processamento da amostra pudessem afetar os resultados. Descobrimos que o indivíduo papua com cobertura quíntupla compartilha 4,0 ± 0,7% mais alelos com o indivíduo Denisova do que o indivíduo francês, e observamos uma assimetria semelhante em todas as 10 comparações de populações melanésias e outras não africanas ( Tabela 1 ). Quando estratificamos os dados por classe de substituição de base e cromossomo, as estatísticas D permanecem qualitativamente inalteradas ( Seção 10 de Informações Suplementares ). Da mesma forma, as estatísticas D observadas ( D são consistentes para todas as profundidades de cobertura de leitura, indicando que erros de mapeamento, por exemplo, devido a duplicações segmentares, provavelmente não explicam esses resultados. Finalmente, diferenças na taxa de erro de sequenciamento entre amostras não podem explicar as estatísticas Seção de Informações Suplementares 10).
Sob a suposição de que o fluxo gênico explica essas observações, determinamos a direção desse fluxo gênico perguntando se os melanésios e outros eurasianos compartilham alelos derivados com os africanos com a mesma frequência. Se o fluxo gênico fosse inteiramente para os ancestrais do indivíduo denisovano, não esperaríamos que isso afetasse o relacionamento dos africanos com os melanésios e outros eurasianos e, portanto, esperaríamos que eles compartilhassem alelos derivados com a mesma frequência com os africanos. No entanto, descobrimos que os alelos derivados em africanos correspondem aos melanésios 3,4 ± 0,4% menos frequentemente do que em outros não africanos ( Z = 10,8). Como essa assimetria é observada sem o uso de dados denisovanos, ela não pode ser explicada pelo fluxo gênico para os denisovanos ou, por exemplo, pela contaminação da amostra denisovana pelo DNA melanésio atual. Assim, pelo menos parte do suposto fluxo gênico deve ter sido para os melanésios ( Informações Suplementares ). seção 8 de
Quando comparamos a assimetria na fração de alelos derivados compartilhados com os dois hominídeos arcaicos com o que seria esperado para indivíduos de ascendência 100% Neandertal ou Denisova, respectivamente ( seção 8 de Informações Suplementares e ref. 8 ), estimamos que 2,5 ± 0,6% dos genomas de populações não africanas derivam de Neandertais, de acordo com nossa estimativa anterior de 1–4%. 8 Além disso, estimamos que 4,8 ± 0,5% dos genomas dos melanésios derivam de denisovanos. No total, até 7,4 ± 0,8% dos genomas dos melanésios podem, portanto, derivar de uma mistura recente com hominídeos arcaicos.
Um modelo de história populacional
Para entender as implicações das relações observadas entre o indivíduo Denisova, os neandertais e os humanos atuais, ajustamos as estatísticas D descritas nas seções anteriores a um modelo parametrizado de história populacional. As estatísticas D para o indivíduo Denisova diferem em dois aspectos importantes daquelas para o neandertal. Primeiro, o indivíduo Denisova compartilha menos alelos derivados com as populações francesa ou chinesa han do que os neandertais. Segundo, o indivíduo Denisova compartilha mais alelos derivados com os papuas do que os neandertais. Somos capazes de ajustar os dados com um modelo que assume que os denisovanos são um grupo irmão dos neandertais com um tempo de divergência populacional de metade a dois terços do tempo para o ancestral comum dos neandertais e humanos. Após a divergência dos denisovanos dos neandertais, houve fluxo gênico dos neandertais para os ancestrais de todos os não africanos atuais. Mais tarde, houve uma miscigenação entre os denisovanos e os ancestrais dos melanésios que não afetou outras populações não africanas. Este modelo é ilustrado em Fig. 3 e é descrito em detalhes na seção 11 de Informações Suplementares .
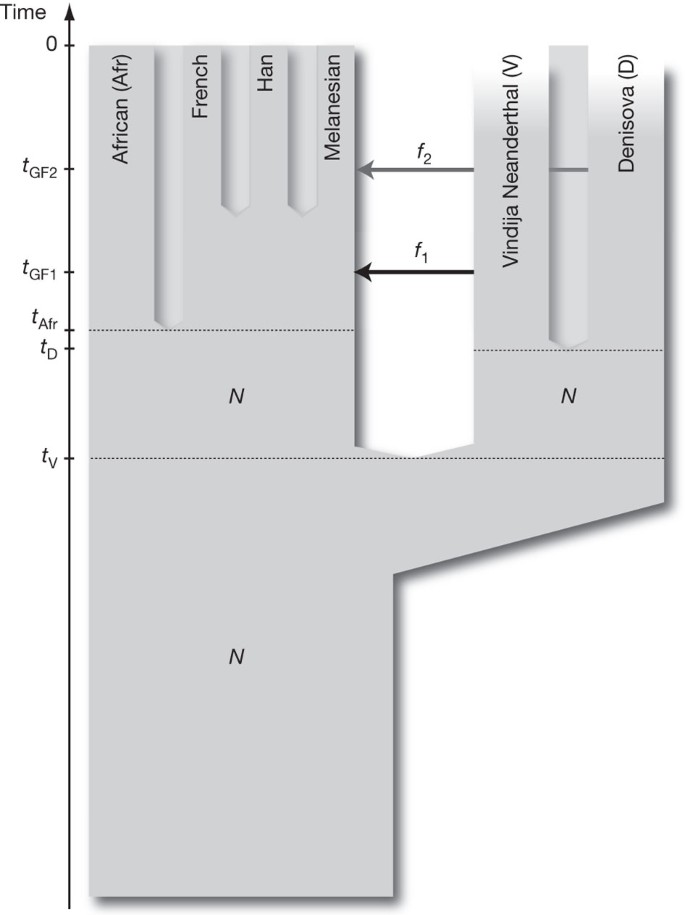
N denotes effective population size, t denotes time of population separation, f denotes amount of gene flow and tGF denotes time of gene flow.
Outros modelos mais complexos também poderiam explicar os dados. Por exemplo, um modelo que invoca apenas o fluxo gênico de Denisovanos para ancestrais Melanésios fora da África e assume quatro subpopulações na África que existiram entre os tempos da origem dos ancestrais Denisovanos e Neandertais e os ancestrais dos atuais Eurasianos também poderia se ajustar aos dados ( Fig. Suplementar 11.4 ). No entanto, como as barreiras ao fluxo gênico entre tais subpopulações teriam que persistir por centenas de milhares de anos para criar os padrões observados, tal modelo é menos plausível em termos biológicos do que um modelo que invoca duas instâncias de fluxo gênico fora da África.
Discordância entre o mtDNA e as histórias nucleares
A história populacional indicada pelo genoma nuclear é diferente daquela indicada pela filogenia do mtDNA. Há duas explicações possíveis para isso. Uma é que a linhagem do mtDNA foi introduzida em ancestrais denisovanos por mistura de outra linhagem de hominídeos para a qual não temos dados. A outra é que a discordância é o resultado de "classificação de linhagem incompleta", isto é, a distribuição aleatória de linhagens genéticas devido à deriva genética, que pode ter permitido que uma linhagem divergente de mtDNA sobrevivesse em denisovanos por acaso, enquanto se perdia em neandertais e humanos modernos. Um grande tamanho populacional ancestral torna a classificação de linhagem incompleta mais provável de ocorrer. Na seção 11 de Informações Suplementares , mostramos que, dadas suposições razoáveis sobre o tamanho das populações ancestrais, a discordância da filogenia do mtDNA com aquela indicada pelo DNA nuclear pode ser explicada por uma pequena quantidade de mistura de outro hominídeo arcaico ou por classificação de linhagem incompleta. Assim, os dados não nos permitem favorecer uma hipótese em detrimento da outra.
Um dente da Caverna Denisova
Em 2000, um dente de hominídeo foi descoberto na camada 11.1 da galeria sul da Caverna Denisova ( Fig. 4a, b ). O dente é de um adulto jovem e, portanto, de um indivíduo diferente da falange, que se origina de um juvenil ( Informações Suplementares, seção 12). Para elucidar a relação do dente com o indivíduo do qual a falange é derivada, extraímos DNA de 50 mg de dentina da raiz do dente e preparamos uma biblioteca de sequenciamento ( Informações Suplementares, seção 13). Cerca de 0,17% das sequências aleatórias de DNA determinadas a partir dessa biblioteca se alinharam ao genoma humano, enquanto o restante provavelmente representa contaminação microbiana comum em ossos antigos. Portanto, utilizamos uma nova abordagem de captura de DNA. 36 para isolar sequências de mtDNA da biblioteca de sequenciamento. Um total de 15.094 sequências foram identificadas, o que permitiu a montagem do genoma completo do mtDNA com uma cobertura média de 58 vezes. Essa sequência difere em duas posições do mtDNA da falange, enquanto difere em cerca de 380 posições tanto do Neandertal quanto dos humanos atuais. O tempo desde o ancestral comum mais recente dos dois mtDNAs da Caverna Denisova é estimado em 7.500 anos, com um limite superior de 95% de 16.000 anos ( seção 13 de Informações Suplementares ). Concluímos que o dente e a falange derivam de dois indivíduos diferentes, provavelmente da mesma população de hominídeos.
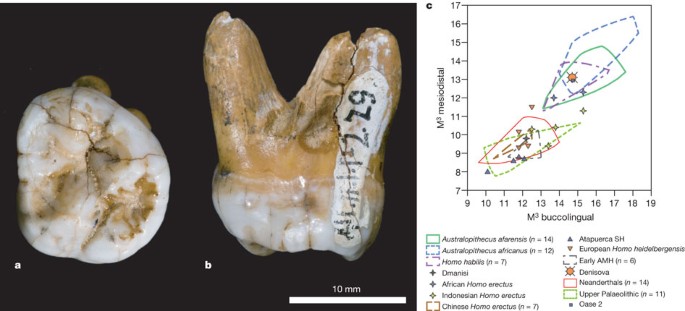
a, b, Occlusal (a) and mesial (b) views. c, Comparison of the Denisova molar to diverse third molars, in a biplot of the mesiodistal and buccolingual lengths (in mm). AMH, anatomically modern humans; SH, Sima de los Huesos. Supplementary Fig. 12.1 presents a similar comparison to second molars.
Morfologia do molar de Denisova
O dente é um molar superior esquerdo quase completo, provavelmente o terceiro, mas possivelmente o segundo ( Fig. 4b ). A coroa é trapezoidal e afunila-se fortemente distalmente, com paredes linguais e vestibulares protuberantes, dando ao dente uma aparência inflada ( Informações Suplementares , seção 12). As raízes são curtas, porém robustas e fortemente alargadas.
No geral, o dente é muito grande (diâmetro mesiodistal, 13,1 mm; bucolingual, 14,7 mm). Como um terceiro molar, está fora da faixa de variação normal de tamanho de todos os táxons fósseis do gênero Homo , com exceção de H. habilis e H. rudolfensis , e comparável aos Australopitecos ( Fig. 4c ). Comparado aos segundos molares, é maior do que os neandertais ou os primeiros humanos modernos, mas semelhante a H. erectus e H. habilis ( Fig. 12.1 suplementar ).
Além do tamanho, também se distingue da maioria dos terceiros molares neandertais pela ausência de redução do hipocone, e dos segundos e terceiros molares neandertais pela presença de uma grande bacia em garra e pelo forte alargamento da coroa. Além disso, não possui a projeção lingual do hipocone observada em todos os primeiros e muitos segundos molares neandertais, e possui raízes fortemente divergentes, ao contrário das raízes pouco espaçadas e frequentemente fundidas dos neandertais.
É de particular interesse comparar o molar de Denisova com os hominídeos do Pleistoceno Médio da China, onde o H. erectus e outras formas arcaicas, às vezes interpretadas como H. heidelbergensis , podem ter sobrevivido até recentemente. Infelizmente, muito poucos desses fósseis preservam terceiros molares superiores. Dos poucos exemplos disponíveis, a maioria difere do molar de Denisova por seu tamanho fortemente reduzido. Os segundos molares são mais frequentes do que os terceiros molares, e a maioria tem formato trapezoidal como o de Denisova, mas não têm a posição lingualmente inclinada do hipocone e do metacone e o forte alargamento basal da coroa.
O molar de Denisova corrobora a evidência de DNA de que a população de Denisova é distinta dos neandertais tardios, bem como dos humanos modernos. De fato, as características primitivas do dente de Denisova sugerem que os denisovanos podem ter sido separados da linhagem neandertal antes que características dentais neandertais fossem documentadas na Eurásia Ocidental (> 300.000 anos a.C. ) ( Informações Suplementares, seção 12), embora não possamos excluir a possibilidade de que a morfologia dentária de Denisova resulte de uma reversão.
Estratigrafia e datação
O pequeno tamanho tanto da falange quanto do dente impede a datação direta por radiocarbono. Em vez disso, datamos sete fragmentos ósseos encontrados próximos aos restos mortais de hominídeos na camada 11 nas galerias leste e sul. Para garantir que estivessem associados à ocupação humana da caverna, escolhemos ossos que apresentam evidências de modificação humana, incluindo uma costela com incisões regulares e um projétil ósseo à queima-roupa, geralmente associado a conjuntos culturais do Paleolítico Superior. Na galeria sul, onde ossos modificados não estavam disponíveis, utilizamos ossos de herbívoros ( Informações Suplementares ). seção 12 de
Quatro das sete datas são infinitas, com mais de 50.000 anos AP (não calibradas), enquanto três são finitas, entre 16.000 e 30.000 anos AP ( Tabela Suplementar 12.1 ). A costela com incisões e o projétil à queima-roupa têm cerca de 30.000 e 23.000 anos AP , respectivamente. Juntamente com três datas anteriores 23 Isso demonstra que a camada 11 contém vestígios culturais de pelo menos dois períodos diferentes: um com mais de 50.000 anos a.C. e outro mais recente. No entanto, a estratigrafia é complicada pela descoberta de uma área em forma de cunha próxima à área onde a falange foi encontrada, que provavelmente está perturbada ( Informações Suplementares , seção 12). Restos de hominídeos grandes o suficiente para permitir datações diretas por radiocarbono podem eventualmente ser descobertos na caverna, mas uma hipótese razoável é que a falange e o molar pertençam à ocupação mais antiga.
Discussion
A preservação molecular da falange de Denisova é excepcional, pois a fração de DNA endógeno em relação ao microbiano é de cerca de 70%. Em contraste, em todos os restos mortais de Neandertais estudados até o momento, a abundância relativa de DNA endógeno é inferior a 5%, e tipicamente inferior a 1%. Além disso, o comprimento médio dos fragmentos de DNA de hominídeos na falange de Denisova é de 58 pares de bases (pb) (SL3003) e 74 pb (SL3004), apesar do tratamento enzimático que remove resíduos de uracila e diminui o tamanho médio do fragmento, enquanto na maioria das amostras de Neandertais bem preservadas, o tamanho é de 50 pb ou menos sem esse tratamento. Assim, embora muitos Neandertais sejam preservados em condições aparentemente semelhantes às da Caverna de Denisova, a falange de Denisova é um dos poucos ossos encontrados em condições temperadas que estão tão bem preservados quanto muitos restos de permafrost. 37 , 38 Não está claro o porquê. Não se deve a alguma condição que afeta todos os restos mortais de hominídeos na Caverna Denisova, pois a fração de DNA endógeno no dente é de 0,17%; ou seja, típica de outros restos mortais de hominídeos do Pleistoceno Superior. É possível que uma rápida dessecação do tecido após a morte, que limitaria a degradação do DNA por enzimas endógenas, bem como o crescimento microbiano, tenha permitido essa preservação excepcional.
O indivíduo Denisova e a população à qual pertencia carregam algumas características moleculares (mtDNA), bem como morfológicas (dentais) excepcionalmente arcaicas. No entanto, o quadro que emerge da análise do genoma nuclear é aquele em que a população Denisova é um grupo irmão dos Neandertais. Três possibilidades poderiam explicar como tais características arcaicas passaram a estar presentes nos Denisovanos. Uma possibilidade é que essas características tenham sido mantidas nos Denisovanos, mas se perderam nos humanos modernos e nos Neandertais. Uma segunda possibilidade, não mutuamente exclusiva, é que elas entraram na população Denisova através do fluxo gênico de algum hominíneo ainda mais divergente. Embora tal fluxo gênico não possa ser detectado com os dados atuais de mtDNA e DNA nuclear, o sequenciamento posterior de outros restos de hominíneos pode, no futuro, permitir testes para sua detecção. Uma terceira possibilidade que poderia explicar a morfologia dentária aparentemente arcaica, mas não o mtDNA, é uma reversão para características ancestrais.
Após a divergência entre si, denisovanos e neandertais tiveram histórias populacionais amplamente distintas, como demonstrado por diversas observações. Primeiro, os padrões de compartilhamento de alelos indicam que os ancestrais denisovanos não contribuíram com genes em um nível detectável para os povos atuais em toda a Eurásia, enquanto os neandertais o fizeram. 8 Assim, os neandertais, em algum momento, interagiram com ancestrais dos atuais eurasianos, independentemente dos denisovanos. Em segundo lugar, a diversidade genética dos neandertais em toda a sua distribuição geográfica nos últimos trinta ou quarenta mil anos de sua história foi extremamente baixa, indicando que eles experimentaram um ou mais gargalos genéticos fortes, independentemente dos denisovanos. Em terceiro lugar, nossos resultados indicam que os denisovanos, mas não os neandertais, contribuíram com genes para os ancestrais dos atuais melanésios. Em quarto lugar, a morfologia dentária não mostra evidências de quaisquer características derivadas observadas em neandertais. De fato, restos dentários da Sima de los Huesos de Atapuerca, para os quais foram propostas idades entre 350.000 e 600.000 anos 39 , 40 , já apresentam características morfológicas semelhantes às do Neandertal que não são vistas no molar de Denisova.
Uma questão interessante é quão difundidos eram os denisovanos. Uma possibilidade é que eles viveram em grandes partes do leste da Ásia na época em que os neandertais estavam presentes na Europa e na Ásia ocidental. Uma observação compatível com essa possibilidade é que parentes denisovanos parecem ter contribuído com genes para os atuais melanésios, mas não para as populações atuais que atualmente vivem muito mais perto da região de Altai, como os chineses han ou os mongóis ( Tabela 1 ). Assim, eles pelo menos em algum momento estiveram presentes em uma área onde interagiram com os ancestrais dos melanésios e isso presumivelmente não foi no sul da Sibéria. Estudos adicionais de características moleculares e morfológicas de restos de hominídeos pela Ásia devem esclarecer quão difundidos eram os denisovanos e como eles estavam relacionados a hominídeos arcaicos que não os neandertais.
O indivíduo Denisova pertence a um grupo de hominídeos que compartilha um ancestral comum com os neandertais, mas possui uma história populacional distinta. Definimos esse grupo com base em evidências genômicas e o chamamos de Denisovanos, mas nos abstemos de quaisquer designações taxonômicas lineanas formais que indiquem o status de espécie ou subespécie para neandertais ou denisovanos. Em nossa opinião, esses resultados mostram que, no continente eurasiano, existiam pelo menos duas formas de hominídeos arcaicos no Pleistoceno Superior: uma forma eurasiana ocidental com características morfológicas comumente usadas para defini-los como neandertais, e uma forma oriental à qual os indivíduos Denisova pertencem. No futuro, quando genomas mais completos desses e de outros hominídeos arcaicos forem sequenciados a partir de restos mortais que permitam a avaliação de mais características morfológicas, suas relações serão ainda mais bem compreendidas. Este será um esforço importante, visto que o quadro emergente da evolução dos hominídeos do Pleistoceno Superior é aquele em que o fluxo gênico entre diferentes grupos de hominídeos era comum.
Resumo dos Métodos
As treze seções das Informações Suplementares fornecem uma descrição completa dos métodos.
Códigos de acesso
Acessos primários
EMBL/GenBank/DDBJ
Depósitos de dados
Os dados de sequência bruta dos dois fósseis de Denisova, dos sete humanos atuais e do mtDNA do dente foram depositados no Arquivo Europeu de Nucleotídeos do EMBL-EBI sob os números de acesso ERP000318 , ERP000121 e FR695060 , respectivamente. Os alinhamentos das leituras de sequência de Denisova com os genomas humano e de chimpanzé estão disponíveis para navegação e download em http://genome.ucsc.edu/Denisova .
Referências
Hublin, JJ A origem dos Neandertais. Processo. Acad. Nacional. Ciência. EUA 106 , 16022–16027 (2009)
Pope, GG Evidências craniofaciais para a origem dos humanos modernos na China. Am. J. Phys. Antropol. 35 (Suppl. 15). 243–298 (1992)
Rightmire, GP Tamanho do cérebro e encefalização em Homo do Pleistoceno Inferior ao Médio. Am. J. Phys. Antropol. 124 , 109–123 (2004)
Wu, X. & Poirier, FE Evolução Humana na Ásia (Oxford Univ. Press, 1995)
Brown, P. et al. A new small-bodied hominin from the Late Pleistocene of Flores, Indonesia. Nature 431, 1055–1061 (2004)
Morwood, MJ et al. Arqueologia e idade de um novo hominídeo de Flores, no leste da Indonésia. Nature 431 , 1087–1091 (2004)
Morwood, MJ et al. Prefácio: pesquisa em Liang Bua, Flores, Indonésia. J. Hum. Evol. 57 , 437–449 (2009)
Green, RE et al. Um rascunho da sequência do genoma neandertal. Science 328 , 710–722 (2010)
Beauval, C. et al. Um fêmur de Neandertal tardio de Les Rochers-de-Villeneuve, França. Processo. Acad. Nacional. Ciência. EUA 102 , 7085–7090 (2005)
Briggs, A. W. et al. Targeted retrieval and analysis of five Neandertal mtDNA genomes. Science 325, 318–321 (2009)
Caramelli, D. et al. A highly divergent mtDNA sequence in a Neandertal individual from Italy. Curr. Biol. 16, R630–R632 (2006)
Green, R. E. et al. A complete Neandertal mitochondrial genome sequence determined by high-throughput sequencing. Cell 134, 416–426 (2008)
Krause, J. et al. Neandertais na Ásia Central e na Sibéria. Nature 449 , 902–904 (2007)
Krings, M. et al. Uma visão da diversidade genética neandertal. Nature Genet. 26 , 144–146 (2000)
Lalueza-Fox, C. et al. DNA mitocondrial de um neandertal ibérico sugere afinidade populacional com outros neandertais europeus. Curr. Biol. 16 , R629–R630 (2006)
Orlando, L. et al. Revisitando a diversidade neandertal com uma sequência de mtDNA de 100.000 anos. Curr. Biol. 16 , R400–R402 (2006)
Serre, D. et al. Nenhuma evidência da contribuição do mtDNA neandertal para os primeiros humanos modernos. PLoS Biol. 2 , e57 (2004)
Endicott, P., Ho, S. Y. & Stringer, C. Using genetic evidence to evaluate four palaeoanthropological hypotheses for the timing of Neanderthal and modern human origins. J. Hum. Evol. 59, 87–95 (2010)
Krause, J. et al. The complete mitochondrial DNA genome of an unknown hominin from southern Siberia. Nature 464, 894–897 (2010)
Derevianko, AP et al. Paleoambiente e ocupação humana paleolítica de Gorny Altai [em russo] (Instituto de Arqueologia e Etnografia SB RAS Press, 2003)
Derevianko, A. O Paleolítico da Sibéria: Novas Descobertas e Interpretações (Univ. Illinois Press, 1998)
Derevianko, AP et al. Arqueologia, Geologia e Paleogeografia do Pleistoceno e Holoceno das Montanhas Altai [em russo] (Nauka, 1998)
Derevianko, AP, Shunkov, MV & Volkov, PV Uma pulseira paleolítica da caverna Denisova. Arqueol. Etnol. Antropol. Eurásia 34 , 13–25 (2008)
Green, RE et al. O genoma neandertal e a autenticidade do DNA antigo. EMBO J. 28 , 2494–2502 (2009)
Rohland, N. & Hofreiter, M. Comparação e otimização da extração de DNA antigo. Biotechniques 42 , 343–352 (2007)
Lindahl, T. et al. DNA N-glicosidases: propriedades da uracila-DNA glicosidase de Escherichia coli . J. Biol. Chem. 252 , 3286–3294 (1977)
Briggs, AW et al. Remoção de citosinas desaminadas e detecção de metilação in vivo em DNA antigo. Nucleic Acids Res. 38 , e87 (2010)
Meyer, M. & Kircher, M. Preparação da biblioteca de sequenciamento Illumina para captura e sequenciamento de alvos altamente multiplexados. Cold Spring Harb. Protoc. 2010. 10.1101/pdb.prot5448 (2010)
Briggs, AW et al. Padrões de danos em sequências de DNA genômico de um neandertal. Proc. Natl Acad. Sci. USA 104 , 14616–14621 (2007)
Kircher, M., Stenzel, U. e Kelso, J. Melhoria na chamada de bases para o Analisador de Genoma Illumina usando estratégias de aprendizado de máquina. Genome Biol. 10 , R83 (2009)
Li, H. & Durbin, R. Alinhamento de leitura curta rápido e preciso com a transformada de Burrows-Wheeler. Bioinformatics 25 , 1754–1760 (2009)
Paten, B. et al. Enredo and Pecan: genome-wide mammalian consistency-based multiple alignment with paralogs. Genome Res. 18, 1814–1828 (2008)
Reich, D. et al. Reduced neutrophil count in people of African descent is due to a regulatory variant in the Duffy antigen receptor for chemokines gene. PLoS Genet. 5, e1000360 (2009)
Keinan, A., Mullikin, JC, Patterson, N. & Reich, D. A medição do espectro de frequência dos alelos humanos demonstra maior deriva genética em asiáticos orientais do que em europeus. Nature Genet. 39 , 1251–1255 (2007)
Li, JZ et al. Relações humanas globais inferidas a partir de padrões de variação em todo o genoma. Science 319 , 1100–1104 (2008)
Maricic, T., Whitten, M. & Pääbo, S. Captura multiplexada de sequências de DNA de genomas mitocondriais usando produtos de PCR. PLoS ONE 5 , e14004 (2010)
Poinar, HN et al. Metagenômica para paleogenômica: sequenciamento em larga escala do DNA de mamutes. Science 311 , 392–394 (2006)
Rasmussen, M. et al. Sequência do genoma humano antigo de um paleoesquimó extinto. Nature 463 , 757–762 (2010)
Bischoff, JL et al. Datações de alta resolução da série U dos hominídeos de Sima de los Huesos rendem 600 +∞ −66 mil anos: Implicações para a Evolução da Linhagem Neandertal Primitiva. J. Archaeol. Sci. 34 , 763–770 (2007)
Bischoff, JL et al. Os hominídeos de Sima de los Huesos datam de além do equilíbrio U/Th (>350 mil anos-luz) e talvez de 400 a 500 mil anos-luz: Novas datações radiométricas. J. Archaeol. Sci. 30 , 275–280 (2003)
Agradecimentos
Agradecemos a C. Bustamante, AI Krivoshapkin, M. Lachmann, R. Nielsen, K. Pruefer, A. Tsybankov, L. Vigilant e W. Zhai pelos comentários; a K. Finstermeier pelo trabalho gráfico; ao grupo de sequenciamento MPI-EVA, R. Schultz e S. Weihnachtsmann pelo suporte técnico; e a P. Fujita, A. Hinrichs e K. Learned pelo desenvolvimento do portal do navegador genômico da UCSC para os dados de Denisova. O Fundo Presidencial de Inovação da Sociedade Max Planck e a Fundação Krekeler forneceram apoio financeiro. MS foi apoiado por uma bolsa dos Institutos Nacionais de Saúde dos EUA (R01-GM40282). A Fundação Nacional de Ciências concedeu uma Bolsa Internacional de Pós-Doutorado (OISE-0754461) a JMG, uma Bolsa em Informática Biológica a PLFJ e uma bolsa HOMINID (1032255) a DR.
Informações do autor
Authors and Affiliations
Contribuições
J.Kr., TM, QF e MM realizaram os experimentos; DR, REG, MK, J.Kr., NP, EYD, AWB, US, PLFJ, TM, JMG, TB-M., CA, SM, HL, EEE, M.St., J.Ke., M.Sl. e SP analisaram dados genéticos; BV, MR, ST, MVS, APD e J.-JH analisaram dados arqueológicos e antropológicos; DR e SP escreveram e editaram o manuscrito.
Autores correspondentes
Declarações de ética
Interesses conflitantes
Os autores declaram não haver interesses financeiros conflitantes.
Informações suplementares
Informações suplementares
Este arquivo contém 13 seções de Informações Suplementares (consulte o Índice), que incluem Dados Suplementares, Figuras Suplementares, Tabelas Suplementares e referências adicionais. Para Dados Suplementares 1, acesse o seguinte link para acessar este arquivo: http://bioinf.eva.mpg.de/download/DenisovaGenome/Denisova_Neandertal_catalog.tgz (PDF 4630 kb)
Supplementary Data 2
This spreadsheet describes the 13 regions of potential admixture from Neandertals into modern humans as described in Green et al., Science 328:710 (2010). (XLS 60 kb)
Direitos e permissões
Este artigo é distribuído sob os termos da licença Creative Commons Atribuição-Uso Não Comercial-Compartilhamento pela mesma Licença ( http://creativecommons.org/licenses/by-nc-sa/3.0/ ), que permite a distribuição e reprodução em qualquer meio, desde que o autor e a fonte originais sejam creditados. Esta licença não permite a exploração comercial, e trabalhos derivados devem ser licenciados sob a mesma licença ou uma licença similar.
Sobre este artigo
Citar este artigo
Reich, D., Green, R., Kircher, M. et al. História genética de um grupo arcaico de hominídeos da Caverna Denisova na Sibéria. Natureza 468 , 1053–1060 (2010). https://doi.org/10.1038/nature09710
Nenhum comentário:
Postar um comentário
Observação: somente um membro deste blog pode postar um comentário.