The tuatara (Sphenodon punctatus)—the only living member of the reptilian order Rhynchocephalia (Sphenodontia), once widespread across Gondwana1,2—is an iconic species that is endemic to New Zealand2,3.
A key link to the now-extinct stem reptiles (from which dinosaurs,
modern reptiles, birds and mammals evolved), the tuatara provides key
insights into the ancestral amniotes2,4.
Here we analyse the genome of the tuatara, which—at approximately
5 Gb—is among the largest of the vertebrate genomes yet assembled. Our
analyses of this genome, along with comparisons with other vertebrate
genomes, reinforce the uniqueness of the tuatara. Phylogenetic analyses
indicate that the tuatara lineage diverged from that of snakes and
lizards around 250 million years ago. This lineage also shows moderate
rates of molecular evolution, with instances of punctuated evolution.
Our genome sequence analysis identifies expansions of proteins,
non-protein-coding RNA families and repeat elements, the latter of which
show an amalgam of reptilian and mammalian features. The sequencing of
the tuatara genome provides a valuable resource for deep comparative
analyses of tetrapods, as well as for tuatara biology and conservation.
Our study also provides important insights into both the technical
challenges and the cultural obligations that are associated with genome
sequencing.
Main
The tuatara is an iconic terrestrial vertebrate that is unique to New Zealand2.
The tuatara is the only living member of the archaic reptilian order
Rhynchocephalia (Sphenodontia), which last shared a common ancestor with
other reptiles at about 250 million years ago (Fig. 1);
this species represents an important link to the now-extinct stem
reptiles from which dinosaurs, modern reptiles, birds and mammals
evolved, and is thus important for our understanding of amniote
evolution2.
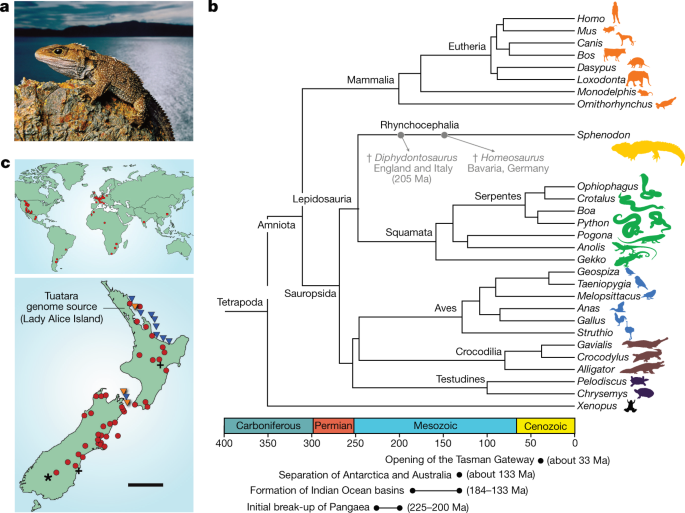
Fig. 1: The phylogenetic significance and distribution of the tuatara.
a, The tuatara, (S. punctatus) is the sole survivor of the order Rhynchocephalia. b, c,
The rhynchocephalians appear to have originated in the early Mesozoic
period (about 250–240 million years ago (Ma)) and were common, speciose
and globally distributed for much of that era. The geographical range of
the rhynchocephalians progressively contracted after the Early Jurassic
epoch (about 200–175 Ma); the most recent fossil record outside of New
Zealand is from Argentina in the Late Cretaceous epoch (about 70 Ma). c,
The last bastions of the rhynchocephalians are 32 islands off the coast
of New Zealand, which have recently been augmented by the establishment
of about 10 new island or mainland sanctuary populations using
translocations. The current global population is estimated to be around
100,000 individuals. Rhynchocephalian and tuatara fossil localities are
redrawn and adapted from ref. 1 with permission, and incorporate data from ref. 2. In the global distribution map (c,
top); triangle = Triassic; square = Jurassic; circle = Cretaceous; and
diamond = Palaeocene. In the map of the New Zealand distribution (c,
bottom); asterisk = Miocene; cross = Pleistocene; circle = Holocene;
blue triangle = extant population; and orange triangle = population
investigated in this study. Scale bar, 200 km. Photograph credit,
F. Lanting.
It is also a species of importance in other contexts. First, the tuatara is a taonga (special treasure) for Māori, who hold that tuatara are the guardians of special places2.
Second, the tuatara is internationally recognized as a critically
important species that is vulnerable to extinction owing to habitat
loss, predation, disease, global warming and other factors2.
Third, the tuatara displays a variety of morphological and
physiological innovations that have puzzled scientists since its first
description2.
These include a unique combination of features that are shared
variously with lizards, turtles and birds, which left its taxonomic
position in doubt for many decades2. This taxonomic conundrum has largely been addressed using molecular approaches4,
but the timing of the split of the tuatara from the lineage that forms
the modern squamates (lizards and snakes), the rate of evolution of
tuatara and the number of species of tuatara remain contentious2.
Finally, there are aspects of tuatara biology that are unique within,
or atypical of, reptiles. These include a unique form of
temperature-dependent sex determination (which sees females produced
below, and males above, 22 °C), extremely low basal metabolic rates and
considerable longevity2.
To provide insights into the biology of the tuatara, we have sequenced its genome in partnership with Ngātiwai, the Māori iwi (tribe) who hold kaitiakitanga
(guardianship) over the tuatara populations located on islands in the
far north of New Zealand. This partnership—which, to our knowledge, is
unique among the genome projects undertaken to date—had a strong
practical focus on developing resources and information that will
improve our understanding of the tuatara and aid in future conservation
efforts. It is hoped that this work will form an exemplar for future
genome initiatives that aspire to meet access and benefit-sharing
obligations to Indigenous communities.
We find that the tuatara
genome—as well as the animal itself—is an amalgam of ancestral and
derived characteristics. Tuatara has 2n = 36 chromosomes in both sexes, consisting of 14 pairs of macrochromosomes and 4 pairs of microchromosomes5.
The genome size, which is estimated to be approximately 5 Gb, is among
the largest of the vertebrate genomes sequenced to date; this is
predominantly explained by an extraordinary diversity of repeat
elements, many of which are unique to the tuatara.
Sequencing, assembly, synteny and annotation
Our tuatara genome assembly is 4.3 Gb, consisting of 16,536 scaffolds with an N50 scaffold length of 3 Mb (Extended Data Table 1, Supplementary Information 1). Genome assessment using Benchmarking Universal Single-Copy Orthologs (BUSCO)6
indicates 86.8% of the vertebrate gene set are present and complete.
Subsequent annotation identified 17,448 genes, of which 16,185 are
one-to-one orthologues (Supplementary Information 2).
Local gene-order conservation is high; 75% or more of tuatara genes
showed conservation with birds, turtles and crocodilians. We also find
that components of the genome, of 15 Mb in size and larger, are syntenic
with other vertebrates; protein-coding gene order and orientation are
maintained between tuatara, turtle, chicken and human, and strong
co-linearity is seen between tuatara contigs and chicken chromosomes
(Extended Data Figs. 1, 2).
Genomic architecture
At
least 64% of the tuatara genome assembly is composed of repetitive
sequences, made up of transposable elements (31%) and low-copy-number
segmental duplications (33%). Although the total transposable element
content is similar to other reptiles7,
the types of repeats we found appear to be more mammal-like than
reptile-like. Furthermore, a number of the repeat families show evidence
of recent activity and greater expansion and diversity than seen in
other vertebrates (Fig. 2).
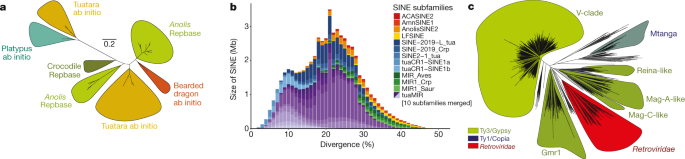
Fig.
2: Analysis of the repeat landscape in the tuatara genome identifies
unique repeat families, evidence of recent activity and a greater
expansion and diversity of repeats than any other amniote.
a,
A phylogenetic analysis on the basis of the reverse transcriptase
domain of L2 repeats identifies two L2 subfamilies; one typical of other
lepidosaurs and one that is similar to platypus L2. This phylogeny is
based on L2 elements >1.5-kb long with a reverse transcriptase domain
of >200 amino acids. b, Landscape plot of SINE
retrotransposons suggests the tuatara genome is dominated by MIR
sequences that are most typically associated with mammals; the tuatara
genome is now the amniote genome in which the greatest MIR diversity has
been observed. Only SINE subfamilies that occupy more than 1,000 bp are
shown. Definitions of the abbreviations of the SINE subfamilies follow:
ACASINE2, Anolis carolinesis SINE family; AmnSINE1, Amniota SINE1; AnolisSINE2, A. carolinesis SINE2 family, LFSINE, lobe-finned fishes SINE; SINE−2019−L_tua, tuatara SINE; SINE-2019_Crp, Crocodylus porosus SINE; SINE2-1_tua, tuatara SINE2; tuaCR1-SINE1a and b, tuatara CR1-mobilized SINEs; MIR_Aves, avian MIR sequence; MIR1_Crp, C. porosus MIR sequence; MIR1_Saur, Sauropsida MIR sequence; tuaMIR, tuatara MIRs. c,
The tuatara genome contains about 7,500 full-length,
long-terminal-repeat retro-elements, including nearly 450 endogenous
retroviruses that span the five major retroviral clades. A Ty1/Copia
element (Mtanga-like) is especially abundant, but Bel-Pao
long-terminal-repeat retro-elements are absent. At least 37 complete
spumaretroviruses are present in the tuatara genome.
L2
elements account for most of the long interspersed elements in the
tuatara genome (10% of the genome), and some may still be active
(Supplementary Information 4). CR1 elements—the dominant long interspersed element in the genomes of other sauropsids8—are rare. CR1 elements comprise only about 4% of the tuatara genome (Fig. 2a, Supplementary Table 4.1), but some are potentially active (Supplementary Fig. 4.4).
L1 elements, which are prevalent in placental mammals, account for only
a tiny fraction of the tuatara genome (<1 a="" data-track-action="supplementary material anchor" data-track-label="link" data-track="click" href="https://www.nature.com/articles/s41586-020-2561-9#MOESM1" table="" upplementary="">4.1
).
However, we find that an L2 subfamily that is present in the tuatara,
but is absent from other lepidosaurs, is also common in monotremes9 (Supplementary Figs. 4.3–4.5).
Collectively, these data suggest that stem-sauropsid ancestors had a
repeat composition that was very different from that inferred in
previous comparisons using mammals, birds and lizards7.
Many
of the short interspersed elements (SINEs) in the tuatara are derived
from ancient common sequence motifs (CORE-SINEs), which are present in
all amniotes10; however, at least 16 SINE subfamilies were recently active in the tuatara genome (Fig. 2b, Supplementary Information 5).
Most of these SINEs are mammalian-wide interspersed repeats (MIRs), and
the diversity of MIR subfamilies in the tuatara is the highest thus far
observed in an amniote11,12. In the human genome, hundreds of fossil MIR elements act as chromatin and regulatory domains13;
the very recent activity of diverse MIR subfamilies in the tuatara
suggests these subfamilies may have influenced regulatory rewiring on
rather recent evolutionary timescales.
We detected 24 newly
identified and unique families of DNA transposon, which suggests
frequent germline infiltration by DNA transposons through horizontal
transfer in the tuatara14.
At least 30 subfamilies of DNA transposon were recently active,
spanning a diverse range of cut-and-paste transposons and polintons
(Supplementary Figs. 5.1, 5.2). This diversity is higher than that found in other amniotes15.
Notably, we found thousands of identical DNA transposon copies, which
suggests very recent—and/or ongoing—activity. Cut-and-paste
transposition probably shapes the tuatara genome, as it does in bats15.
We
identified about 7,500 full-length, long-terminal-repeat retro-elements
(including endogenous retroviruses), which we classified into 12 groups
(Fig. 2c, Supplementary Information 6). The general spectrum of long-terminal-repeat retroelements in the tuatara is comparable to that of other sauropsids7,15. We found at least 37 complete spumaretroviruses, which are among the most ancient of endogenous retroviruses16, in the tuatara genome (Fig. 2c, Supplementary Figs. 6.1, 6.2).
The
tuatara genome contains more than 8,000 elements related to non-coding
RNA. Most of these elements (about 6,900) derive from recently active
transposable elements, and overlap with a newly identified CR1-mobilized
SINE (Fig. 2b, Supplementary Information 7).
The remaining high-copy-number elements are sequences closely related
to ribosomal RNAs, spliceosomal RNAs and signal-recognition particle
RNAs.
Finally, a high proportion (33%) of the tuatara genome
originates from low-copy-number segmental duplications; 6.7% of these
duplications are of recent origin (on the basis of their high level of
sequence identity (>94% identity)), which is more than seen in other
vertebrates9.
The tuatara genome is 2.4× larger than the anole genome, and this
difference appears to be driven disproportionately by segmental
duplications.
Overall, the repeat architecture of the tuatara
is—to our knowledge—unlike anything previously reported, showing a
unique amalgam of features that have previously been viewed as
characteristic of either reptilian or mammalian lineages. This
combination of ancient amniote features—as well as a dynamic and diverse
repertoire of lineage-specific transposable elements—strongly reflects
the phylogenetic position of this evolutionary relic.
Our low-coverage bisulfite-sequencing analysis found approximately 81% of CpG sites are methylated in tuatara (Fig. 3a)—the
highest reported percentage of methylation for an amniote. This pattern
differs from that observed in mouse, human (about 70%) and chicken
(about 50%), and is more similar to that of Xenopus (82%) and
zebrafish (78%). One possible explanation for this high level of DNA
methylation is the large number of repetitive elements found in tuatara,
many of which appear recently active and might be regulated via DNA
methylation.
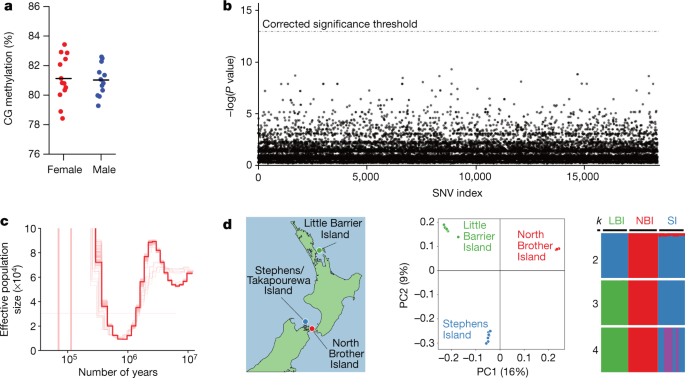
Fig. 3: Analysis of sex differences, demographic history and population structure.
a, Methylation levels in the tuatara genome are high (mean 81%), but show no significant differences among the sexes (female n = 13, mean = 81.13, s.d. = 1.55; male n = 12; mean = 81.02, s.d.−1.07). The black horizontal line represents the mean in each dataset. b,
No single-nucleotide variant (SNV) is significantly differentiated with
respect to sex in the tuatara genome. Each point represents a P value
from a test of sexual differentiation for a single SNV. The dashed line
represents the threshold for statistical significance after accounting
for multiple testing (n = 28; 13 males and 15 females). P values calculated using Fisher’s exact test, two-tailed test and corrected for multiple testing using the Bonferroni method. c, Pairwise sequential Markovian coalescent plot of the demographic history of tuatara using a mutation rate of 1.4 × 10−8 substitutions per site per generation and a generation time of 30 years. d, We examined the three known axes of genetic diversity in tuatara: northern New Zealand (Little Barrier Island (LBI) (n = 9)) and two islands in the Cook Strait (Stephens Island (SI) (n = 9) and North Brother Island (NBI) (n = 10)),
using genotype-by-sequencing methods. Principal component (PC) analysis
and structure plots demonstrate substantial structure among tuatara
populations, and strongly support previous suggestions that the tuatara
on the North Brother Island are genetically distinct and warrant
separate management.
The low normalized CpG content of the tuatara suggests its genome has endured substantial historic methylation17. The tuatara has a significantly bimodal distribution of normalized CpG (Extended Data Fig. 3)
in all of the genomic regions we examined, a similarity it shares with
other reptiles that have temperature-dependent sex determination17.
The low normalized CpG count of the tuatara in non-promoter regions may
result from methylation silencing of repeat elements, and the
bimodality of normalized CpG promoters suggests dual transcriptional
regulation (Extended Data Fig. 3, Supplementary Information 8).
The
mitochondrial genome in the tuatara reference animal is 18,078 bp in
size, containing 13 protein-coding, 2 ribosomal RNA and 22 transfer
(t)RNA genes, a gene content typical among animals (Extended Data Fig. 4). This contradicts previous reports18 that the tuatara mitochondrial genome lacks three genes: ND5, tRNAThr and tRNAHis. These genes are found—with an additional copy of tRNALeu(CUN)
and an additional non-coding block (which we refer to as NC2)—in a
single segment of the mitochondrial genome. Three non-coding areas (NC1,
NC2 and NC3) with control-region (heavy-strand replication origin)
features, and two copies of tRNALeu(CUN)
adjacent to NC1 and NC2, possess identical or near-identical sequences
that are unique to the tuatara mitochondrial genome. These three
non-coding regions may be a result of concerted evolution.
Genomic innovations
As
befits the taxonomic distinctiveness of the tuatara, we find that its
genome displays multiple innovations in genes that are associated with
immunity, odour reception, thermal regulation and selenium metabolism.
Genes
of the major histocompatibility complex (MHC) have an important role in
disease resistance, mate choice and kin recognition, and are among the
most polymorphic genes in the vertebrate genome. Our annotation of MHC
regions in the tuatara, and comparisons of the gene organization with
that of six other species, identified 56 MHC genes (Extended Data Fig. 5, Supplementary Information 9).
Of
the six comparison species, the genomic organization of tuatara MHC
genes is most similar to that of the green anole, which we interpret as
typical for Lepidosauria. Tuatara and other reptiles show a gene content
and complexity more similar to the MHC regions of amphibians and
mammals than to the highly reduced MHC of birds. Although the majority
of genes annotated in the tuatara MHC are well-conserved as one-to-one
orthologues, we observed extensive genomic rearrangements among these
distant lineages.
The tuatara is a highly visual predator that is able to capture prey under conditions of extremely low light2.
Despite the nocturnal visual adaptation of the tuatara, it shows strong
morphological evidence of an ancestrally diurnal visual system19. We identified all five of the vertebrate visual opsin genes in the tuatara genome (Supplementary Information 10).
Our
comparative analysis revealed one of the lowest rates of visual-gene
loss known for any amniote, which contrasts sharply with the high rates
of gene loss observed in ancestrally nocturnal lineages (Extended Data
Fig. 6).
Visual genes involved in phototransduction showed strong negative
selection and no evidence for the long-term shifts in selective
pressures that have been observed in other groups with evolutionarily
modified photoreceptors20.
The retention of five visual opsins and the conserved nature of the
visual system also suggests tuatara possess robust colour vision,
potentially at low light levels. This broad visual repertoire may be
explained by the dichotomy in tuatara life history: juvenile tuatara
often take up a diurnal and arboreal lifestyle to avoid the terrestrial,
nocturnal adults that may predate them2. Collectively, these results suggest a unique path to nocturnal adaptation in tuatara from a diurnal ancestor.
Odorant
receptors are expressed in the dendritic membranes of olfactory
receptor neurons and enable the detection of odours. Species that depend
strongly on their sense of smell to interact with their environment,
find prey, identify kin and avoid predators may be expected to have a
large number of odorant receptors. The tuatara genome contains
472 predicted odorant receptors, of which 341 sequences appear intact
(Supplementary Information 11).
The remainder lack the initial start codon, have frameshifts or are
presumed to be pseudogenes. Many odorant receptors were found as tandem
arrays, with up to 26 genes found on a single scaffold.
The number
and diversity of odorant receptor genes varies greatly in Sauropsida:
birds have 182–688 such genes, the green anole lizard has 156 genes, and
crocodilians and testudines have 1,000–2,000 genes21.
The tuatara has a number of odorant receptors similar to that of birds,
but contains a high percentage of intact odorant receptor genes (85%)
relative to published odorant receptor sets from the genomes of other
sauropsids. This may reflect a strong reliance on olfaction by tuatara,
and therefore pressure to maintain a substantial repertoire of odorant
receptors (Extended Data Fig. 7). There is some evidence that olfaction has a role in identifying prey2, as well as suggestions that cloacal secretions may act as chemical signals.
The
tuatara is a behavioural thermoregulator, and is notable for having the
lowest optimal body temperature of any reptile (16–21 °C). Genes that
encode transient receptor potential ion channels (TRP genes) have an
important role in thermoregulation, as these channels participate in
thermosensation and cardiovascular physiology22;
this led us to hypothesize that TRP genes may be linked to the thermal
tolerance of the tuatara. Our comparative genomic analysis of TRP genes
in the tuatara genome identified 37 TRP-like sequences, spanning all
7 known subfamilies of TRP genes (Extended Data Fig. 8, Supplementary Information 12)— an unusually large repertoire of TRP genes.
Among
this suite of genes, we identified thermosensitive and
non-thermosensitive TRP genes that appear to result from gene
duplication, and have been differentially retained in the tuatara. For
example, the tuatara is unusual in possessing an additional copy of a
thermosensitive TRPV-like gene (TRPV1/2/4, sister to the genes TRPV1, TRPV2 and TRPV4) that has classically been linked to the detection of moderate-to-extreme heat22—a feature it shares with turtles. A strong signature of positive selection among heat-sensitive TRP genes (TRPA1, TRPM and TRPV) was also observed.
In
general, these results show a high rate of differential retention and
positive selection in genes for which a function in heat sensation is
well-established22.
It therefore seems probable that the genomic changes in TRP genes are
associated with the evolution of thermoregulation in tuatara.
Barring tortoises, tuatara are the longest lived of the reptiles—probably exceeding 100 years of age2.
This enhanced lifespan may be linked to genes that afford protection
against reactive oxygen species. One class of gene products that affords
such protection is the selenoproteins. The human genome encodes
25 selenoproteins, the roles of which include antioxidation, redox
regulation, thyroid hormone synthesis and calcium signal transduction,
among others23.
We
identified 26 genes that encode selenoproteins in the tuatara genome,
as well as 4 selenocysteine-specific tRNA genes; all of these appear to
be functional (Supplementary Information 13).
Although further work is needed, the additional selenoprotein gene
(relative to the human genome) and the selenocysteine-specific tRNA
genes may be linked to the longevity of tuatara or might have arisen as a
response to the low levels of selenium and other trace elements in the
terrestrial systems of New Zealand.
Tuatara has a unique mode of
temperature-dependent sex determination, in which higher temperatures
during egg incubation result in males2. We found orthologues for many genes that are known to act antagonistically in masculinizing (for example, SF1 and SOX9) and feminizing (for example, RSPO1 and WNT4) gene networks to promote testicular or ovarian development, respectively24.
We also found orthologues of several genes that have recently been
implicated in temperature-dependent sex determination, including CIRBP24 (Supplementary Information 17, Supplementary Table 17.2). Tuatara possess no obviously differentiable sex chromosomes5, and we found no significant sex-specific differences in global CG methylation (Fig. 3a) and no sex-specific single-nucleotide variants between male and female tuatara (Fig. 3b).
On a gene-by-gene basis, sex-specific differences in methylation and
gene expression patterns probably exist, but this remains to be
investigated.
Phylogeny and evolutionary rates
Our
phylogenomic analyses, which incorporated both whole-genome alignments
and clusters of single-copy orthologues (Supplementary Information 14, 15) recapitulated many patterns that have been observed in the fossil record and corroborated during the genomic era (Fig. 1). After their appearance about 312 million years ago25,
amniote vertebrates diversified into two groups: the synapsids (which
include all mammals) and the sauropsids (which include all reptiles and
birds). We obtained full phylogenomic support for a monophyletic
Lepidosauria, marked by the divergence of the tuatara lineage from all
squamates (lizards and snakes) during the early part of the Triassic
period at about 250 million years ago, as estimated using a penalized
likelihood method (Fig. 1, Supplementary Information 14–16).
The
rate of molecular evolution in the tuatara has previously been
suggested to be paradoxically high, in contrast to the apparently slow
rate of morphological evolution26,27.
However, we find that the actual divergence in terms of DNA
substitutions per site per million years at fourfold degenerate sites is
relatively low, particularly with respect to lizards and snakes; this
makes the tuatara the slowest-evolving lepidosaur yet analysed (Extended
Data Fig. 9a, b).
We also find that in general amniote evolution can be described by a
model of punctuated evolution, in which the amount of genomic change is
related to the degree of species diversification within clades28,29.
The tuatara falls well below this trend, accumulating substitutions at a
rate expected given the lack of rhynchocephalian diversity (Extended
Data Fig. 9c, Supplementary Information 16). This suggests that rates of phenotypic and molecular evolution were not decoupled throughout the evolution of amniotes30.
Patterns of selection
In
two sets of analyses, we find that most genes exhibit a pattern of
molecular evolution that suggests that the tuatara branch evolves at a
different rate than the rest of the tree (Supplementary Information 17, Supplementary Table 4). Approximately 659 of the 4,284 orthologues we tested had significantly different ω
values (ratios of non-synonymous to synonymous substitutions, dN/dS) on
the tuatara branch relative to the birds and other reptiles we tested
(Supplementary Information 17). Although none of these orthologues had ω
values suggestive of strong positive selection (that is, >1), the
results do indicate that shifts in patterns of selection are affecting
many genes and functional categories of genes across the tuatara genome,
including genes involved in RNA regulation, metabolic pathways, general
metabolism and sex determination.
Population genomics
Once
widespread across the supercontinent of Gondwana, Rhynchocephalia is
now represented by a single species (the tuatara) found on a few islands
offshore of New Zealand (Fig. 1c). Historically, tuatara declined in range and numbers because of introduced pests and habitat loss2.
They remain imperilled owing to their highly restricted distribution,
threats imposed by disease and changes in sex ratios induced by climate
change that could markedly affect their survival31.
Previous work has found that populations in northern New Zealand are
genetically distinct from those in the Cook Strait, and that the
population on North Brother Island in the Cook Strait might be a
distinct species3. Although subsequent studies have not supported species status for the population on North Brother Island32, it is managed as a separate conservation unit.
We
used the tuatara reference genome to perform ancestral demographic and
population genomic analyses of this species. First, we investigated
genome-wide signals for demographic change using a pairwise sequentially
Markovian coalescent method (Supplementary Information 18). Our reconstructed demography (Fig. 3c) reveals an increase in effective population size (Ne) that is detectable around 10 million years ago, a marked decrease in Ne about 1–3 million years ago and a rapid increase in Ne
between 500 thousand million years ago and 1 million years ago. These
events correlate well with the known geological history of New Zealand33,
and may reflect an increase in available landmass subsequent to
Oligocene drowning, a period of considerable climatic cooling that
probably reduced tuatara habitat and the formation of land bridges that
facilitated population expansion.
Our population genomic analyses examined the major axes of genetic diversity in tuatara32,34, and revealed substantial genetic structure (Fig. 3d, Supplementary Information 19). Our genome-wide estimate of the fixation index (FST)
is 0.45, and more than two-thirds of variable sites have an allele that
is restricted to a single island. All populations have relatively low
genetic diversity (nucleotide diversity ranges from 8 × 10−4 for North Brother Island to 1.1 × 10−3
for Little Barrier Island). The low within-population diversity and
marked population structure we observe in the tuatara suggests that the
modern island populations were isolated from each other sometime during
the Last Glacial Maximum at about 18 thousand years ago.
Our results also support the distinctiveness of the North Brother Island tuatara, which has variously been described as S. punctatus or Sphenodon guntheri3,32.
This population is highly inbred and shows evidence of a severe
bottleneck, which most probably reflects a founder event around the time
of the last glaciation34.
It is not clear whether the distinctiveness we observe is due to
changes in allele frequency brought about by this bottleneck, or is
reflective of a deeper split in the population history of tuatara.
Regardless, this population is an important source of genetic diversity
in tuatara, possessing 8,480 private alleles. Although we support
synonymization of S. punctatus and S. guntheri32, the ongoing conservation of the North Brother Island population as an independent unit is recommended.
A cultural dimension
The tuatara is a taonga for many Māori—notably Ngātiwai and Ngāti Koata who are the kaitiaki (guardians) of tuatara. We worked in partnership with Ngātiwai iwi
to increase knowledge and understanding of tuatara, and aid in the
conservation of this species in the long term. Ngātiwai were involved in
all decision-making regarding the use of the genome data by potential
collaborators; for each new project we proposed, we discussed the
benefits that might accrue from this work and how these could be shared.
The need to engage with—and protect the rights of—Indigenous
communities in such a transparent way has seldom been considered in the
genome projects published to date, but is a mandated consideration under
the Nagoya Protocol (https://www.cbd.int/abs/).
Our partnership is a step towards an inclusive model of genomic
science, which we hope others will adopt and improve upon. Although each
partnership is unique, we provide a template agreement (Supplementary
Information 20) that we hope will be useful to others.
Discussion
The
tuatara has a genomic architecture unlike anything previously reported,
with an amalgam of features that have previously been viewed as
characteristic of either mammals or reptiles. Notable among these
features are unusually high levels of repetitive sequences that have
traditionally been considered mammalian, many of which appear to have
been recently active, and—to our knowledge—the highest level of genome
methylation thus far reported. We also found a mitochondrial-genome gene
content at odds with previously published reports that omitted the ND5 gene18; this gene is present, nested within a repeat-rich region of the mitochondrial DNA.
Our
phylogenetic studies provide insights into the timing and speed of
amniote evolution, including evidence of punctuated genome evolution
across this phylogeny. We also find that, in contrast to previous
suggestions that the evolutionary rate for tuatara is exceptionally fast26, it is the slowest-evolving lepidosaur yet analysed.
Our
investigations of genomic innovations identified genetic candidates
that may explain the ultra-low active body temperature, longevity and
apparent resistance to infectious disease in tuatara. Further functional
exploration will refine our understanding of these unusual facets of
tuatara biology, and the tuatara genome itself will enable many future
studies to explore the evolution of complex systems across the
vertebrates in a more complete way than has previously been possible.
Our
population genomic work reveals considerable genetic differences among
tuatara populations, and supports the distinctiveness of the North
Brother Island tuatara.
Finally, this genome will greatly aid in
future work on population differentiation, inbreeding and local
adaptation in this global icon, the last remaining species of the once
globally dominant reptilian order Rhynchocephalia.
Methods
No
statistical methods were used to predetermine sample size. The
experiments were not randomized and investigators were not blinded to
allocation during experiments and outcome assessment.
A full description of the methods can be found in the Supplementary Information.
Sampling and sequencing
A
blood sample was obtained from a large male tuatara from Lady Alice
Island (35° 53′ 24.4′′ S 174° 43′ 38.2′′ E) (New Zealand), with
appropriate ethical permissions and iwi consultation and support (Supplementary Information 20).
Total genomic DNA and RNA were extracted and sequenced using the
Illumina HiSeq 2000 and MiSeq sequencing platforms (Illumina) supported
by New Zealand Genomics (Supplementary Information 1).
Genome, transcriptome and epigenome
Raw
reads were de novo-assembled using Allpaths-LG (version 49856). With a
total input data of 5,741,034,516 reads for the paired-end libraries and
2,320,886,248 reads of the mate-pair libraries, our optimal assembly
used 85% of the fragment libraries and 100% of the jumping libraries
(Supplementary Information 1.4). We further scaffolded the assembly using Chicago libraries and HiRise (Supplementary Information 1.3).
We
assembled a de novo transcriptome as a reference for read-mapping using
total RNA derived from the blood of our reference male tuatara, and a
collection of transcriptomic data previously collected from early-stage
embryos35.
In total, we had 131,580,633 new 100-bp read pairs and 60,637,100
previous 50-bp read pairs. These were assembled using Trinity v.2.2.0
(Supplementary Information 1.4).
Low-coverage
bisulfite sequencing was undertaken using a modified post-bisulfite
adaptor tagging method to explore global patterns of methylation in the
genome for 12 male and 13 female tuatara (Fig. 3d, Supplementary Information 1.5).
Repeat and gene annotation
We
used a combination of ab initio repeat identification in
CARP/RepeatModeler/LTRharvest, manual curation of specific newly
identified repeats, and homology to repeat databases to investigate the
repeat content of the tuatara genome (Supplementary Information 1.6).
From these three complementary repeat identification approaches, the
CARP results were in-depth-annotated for long interspersed elements and
segmental duplications (Supplementary Information 4), the RepeatModeler results were in-depth-annotated for SINEs and DNA transposons (Supplementary Information 5), and the LTRharvest results were in-depth-annotated for long-terminal-repeat retrotransposons (Supplementary Information 6).
For
the gene annotation, we used RepeatMasker (v.4.0.3) along with our
partially curated RepeatModeler library plus the Repbase sauropsid
repeat database to mask transposable elements in the genome sequence
before the gene annotation. We did not mask simple repeats at this point
to allow for more efficient mapping during the homology-based step in
the annotation process. Simple repeats were later soft-masked and
protein-coding genes predicted using MAKER2. We used anole lizard (A. carolinensis, version AnoCar2.0), python (Python bivittatus, version bivittatus-5.0.2) and RefSeq (www.ncbi.nlm.nih.gov/refseq)
as protein homology evidence, which we integrated with ab initio gene
prediction methods including BLASTX, SNAP and Augustus. Non-coding RNAs
were annotated using Rfam covariance models (v.13.0) (Supplementary
Information 7).
Orthologue calling
We
performed a phylogenetic analysis to infer orthology relationships
between the tuatara and 25 other species using the Ensembl GeneTree
method (Supplementary Information Tables 2.1, 2.2). Multiple-sequence alignments, phylogenetic trees and homology relationships were extracted in various formats (https://zenodo.org/record/2542571).
We also calculated the gene order conservation score, which uses local
synteny information around a pair of orthologous genes to compute how
much the gene order is conserved. For each of these species, we chose
the paralogue with the best gene order conservation score and sequence
similarity, which resulted in a total set of 3,168 clusters of
orthologues (Supplementary Information 2, Table 2.3).
Gene tree reconstructions and substitution rate estimation
We
constructed phylogenies using only fourfold-degenerate-site data
derived from whole-genome alignments for 27 tetrapods, analysed as a
single partition in RAxML v.8.2.3. Using the topology and branch lengths
obtained from the best maximum likelihood phylogeny, we estimated
absolute rates of molecular evolution in terms of substitution per site
per million years and estimated the divergence times of amniotes via the
semiparametric penalized likelihood method in r8s v.1.8 (Supplementary
Information 14.5).
We
also generated gene trees on the basis of 245 single-copy orthologues
found between all species using a maximum-likelihood-based multi-gene
approach (Supplementary Information 15).
Sequences were aligned using the codon-based aligner PRANK. The FASTA
format alignments were then converted to PHYLIP using the
catfasta2phyml.pl script (https://github.com/nylander/catfasta2phyml).
Next, we used the individual exon PHYLIP files for gene tree
reconstruction using RaxML using a GTR + G model. Subsequently, we
binned all gene trees to reconstruct a species tree and carried out
bootstrapping using Astral (Supplementary Information 15, Supplementary Fig. 15.1).
Divergence times and tests of punctuated evolution
We
inferred time-calibrated phylogenies with BEAST v.2.4.8 using the
CIPRES Science Gateway to explore divergence times (Supplementary
Information 16.1).
We then used Bayesian phylogenetic generalized least squares to regress
the total phylogenetic path length (of fourfold-degenerate sites) on
the net number of speciation events (nodes in a phylogenetic tree) as a
test for punctuated evolution (Supplementary Information 16.2).
Analysis of genomic innovations
We
explored the genomic organization and sequence evolution of genes
associated with immunity, vision, smell, thermoregulation, longevity and
sex determination (Supplementary Information 8–13).
Tests of selection were undertaken across multiple genes, including
those linked to metabolism, vision and sex determination using
multispecies alignments and PAML (Supplementary Information 17).
Population genomics
Demographic
history was inferred from the diploid sequence of our tuatara genome
using a pairwise sequential Markovian coalescent method (Supplementary
Information 18).
We also sampled 10 tuatara from each of three populations that span the
main axes of genetic diversity in tuatara (Supplementary Information 19, Table 19.1),
and used a modified genotype-by-sequencing approach to obtain the SNVs
that we used for population genomic analysis, investigations of loci
associated with sexual phenotype and estimates of genetic load
(Supplementary Information 19).
Permits and ethics
This project was undertaken in partnership with Ngatiwai and in consultation with other iwi who are kaitiaki of tuatara (Supplementary Information 20).
Samples were collected under Victoria University of Wellington Animal
Ethics approvals 2006R12; 2009R12; 2012R33; 22347 and held and used
under permits 45462-DOA and 32037-RES 32037-RES issued by the New
Zealand Department of Conservation.
The Tuatara Genome Consortium Project whole-genome shotgun and genome assembly are registered under the umbrella BioProjects PRJNA418887 and PRJNA445603, which are associated with BioSamples SAMN08038466 and SAMN08793959. Transcriptome read data are submitted under SRR7084910 (whole blood), together with previous data (SRR485948). The transcriptome assembly is submitted to GenBank with ID GGNQ00000000.1.
Illumina short-read, Oxford Nanopore and PacBio long-read sequences are
in the Sequence Read Archive accessions associated with PRJNA445603.
The genome assembly (GCA_003113815.1) described in this paper is
version QEPC00000000.1 and consists of sequences
QEPC01000001–QEPC01016536. Maker gene predictions are available from
Zenodo at https://doi.org/10.5281/zenodo.1489353. The repeat library database developed for tuatara is available from Zenodo at https://doi.org/10.5281/zenodo.2585367.
References
1.
Jones,
M. E., Tennyson, A. J., Worthy, J. P., Evans, S. E. & Worthy, T. H.
A sphenodontine (Rhynchocephalia) from the Miocene of New Zealand and
palaeobiogeography of the tuatara (Sphenodon). Proc. R. Soc. Lond. B276, 1385–1390 (2009).
Cree, A. Tuatara: Biology and Conservation of a Venerable Survivor (Canterbury Univ. Press, 2014).
3.
Daugherty, C. H., Cree, A., Hay, J. M. & Thompson, M. B. Neglected taxonomy and continuing extinctions of tuatara (Sphenodon). Nature347, 177–179 (1990).
Jones,
M. E. H. et al. Integration of molecules and new fossils supports a
Triassic origin for Lepidosauria (lizards, snakes, and tuatara). BMC Evol. Biol. 13, 208 (2013).
O’Meally, D., Miller, H., Patel, H. R., Graves, J. A. M. & Ezaz, T. The first cytogenetic map of the tuatara, Sphenodon punctatus. Cytogenet. Genome Res. 127, 213–223 (2009).
Simão,
F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. &
Zdobnov, E. M. BUSCO: assessing genome assembly and annotation
completeness with single-copy orthologs. Bioinformatics31, 3210–3212 (2015).
Pasquesi, G. I. M. et al. Squamate reptiles challenge paradigms of genomic repeat element evolution set by birds and mammals. Nat. Commun. 9, 2774 (2018).
Zeng,
L., Kortschak, R. D., Raison, J. M., Bertozzi, T. & Adelson, D. L.
Superior ab initio identification, annotation and characterisation of
TEs and segmental duplications from genome assemblies. PLoS ONE13, e0193588 (2018).
Gilbert, N. & Labuda, D. CORE-SINEs: eukaryotic short interspersed retroposing elements with common sequence motifs. Proc. Natl Acad. Sci. USA96, 2869–2874 (1999).
Schaack,
S., Gilbert, C. & Feschotte, C. Promiscuous DNA: horizontal
transfer of transposable elements and why it matters for eukaryotic
evolution. Trends Ecol. Evol. 25, 537–546 (2010).
Sotero-Caio,
C. G., Platt, R. N. II, Suh, A. & Ray, D. A. Evolution and
diversity of transposable elements in vertebrate genomes. Genome Biol. Evol. 9, 161–177 (2017).
Radhakrishnan,
S., Literman, R., Mizoguchi, B. & Valenzuela, N. MeDIP-seq and nCpG
analyses illuminate sexually dimorphic methylation of gonadal
development genes with high historic methylation in turtle hatchlings
with temperature-dependent sex determination. Epigenetics Chromatin10, 28 (2017).
Rest, J. S. et al. Molecular systematics of primary reptilian lineages and the tuatara mitochondrial genome. Mol. Phylogenet. Evol. 29, 289–297 (2003).
Meyer-Rochow, V. B., Wohlfahrt, S. & Ahnelt, P. K. Photoreceptor cell types in the retina of the tuatara (Sphenodon punctatus) have cone characteristics. Micron36, 423–428 (2005).
Schott,
R. K., Bhattacharyya, N. & Chang, B. S. W. Evolutionary signatures
of photoreceptor transmutation in geckos reveal potential adaptation and
convergence with snakes. Evolution73, 1958–1971 (2019).
Vandewege,
M. W. et al. Contrasting patterns of evolutionary diversification in
the olfactory repertoires of reptile and bird genomes. Genome Biol. Evol. 8, 470–480 (2016).
Blair Hedges, S. & Kumar, S. The Timetree of Life (Oxford Univ. Press, 2009).
26.
Hay, J. M., Subramanian, S., Millar, C. D., Mohandesan, E. & Lambert, D. M. Rapid molecular evolution in a living fossil. Trends Genet. 24, 106–109 (2008).
Miller, H. C., Moore, J. A., Allendorf, F. W. & Daugherty, C. H. The evolutionary rate of tuatara revisited. Trends Genet. 25, 13–15, author reply 16–18 (2009).
Subramanian,
S., Hay, J. M., Mohandesan, E., Millar, C. D. & Lambert, D. M.
Molecular and morphological evolution in tuatara are decoupled. Trends Genet. 25, 16–18 (2009).
Mitchell,
N. J., Kearney, M. R., Nelson, N. J. & Porter, W. P. Predicting the
fate of a living fossil: how will global warming affect sex
determination and hatching phenology in tuatara? Proc. R. Soc. Lond. B275, 2185–2193 (2008).
Hay,
J. M., Sarre, S. D., Lambert, D. M., Allendorf, F. W. & Daugherty,
C. H. Genetic diversity and taxonomy: a reassessment of species
designation in tuatara (Sphenodon: Reptilia). Conserv. Genet. 11, 1063–1081 (2010).
Cooper, A. & Cooper, R. A. The Oligocene bottleneck and New Zealand biota: genetic record of a past environmental crisis. Proc. R. Soc. Lond. B261, 293–302 (1995).
MacAvoy, E. S. et al. Genetic variation in island populations of tuatara (Sphenodon spp) inferred from microsatellite markers. Conserv. Genet. 8, 305–318 (2007).
Miller,
H. C., Biggs, P. J., Voelckel, C. & Nelson, N. J. De novo sequence
assembly and characterisation of a partial transcriptome for an
evolutionarily distinct reptile, the tuatara (Sphenodon punctatus). BMC Genomics13, 439 (2012).
N.J.G. acknowledges the support of Ngatiwai iwi,
Allan Wilson Centre, University of Otago, New Zealand Department of
Conservation, New Zealand Genomics and Illumina. J.I.A. was supported by
CONICYT National Doctoral Scholarship No. 21130515. M.W. was supported
by NIH grant R35 GM124827. Ensembl annotation was supported by the
Wellcome Trust (WT108749/Z/15/Z) and the European Molecular Biology
Laboratory. We thank Ngāti Koata, Te Ātiawa o Te Waka-a-Māui, and Ngāti
Manuhiri iwi for granting permission to reuse tuatara samples
obtained from Stephens Island (Takapourewa), North Brother Island and
Little Barrier Island (Hauturu), respectively; all of the people
involved in obtaining and curating the samples held in the Victoria
University of Wellington tuatara collection; A. Zimin, D. Puiu, G.
Marcais, J. Yorke and R. Crowhurst for help with and discussions about
genome assembly; I. Fiddes, J. Armstrong and B. Paten for help with
comparative genome alignments and annotation; the National eScience
Infrastructure (NeSI) and Swedish National Infrastructure for Computing
(SNIC) through the Uppsala Multidisciplinary Center for Advanced
Computational Science (UPPMAX) for computational support; R. McPhee for
help with figures; and T. Braisher for manuscript coordination and
editing.
Author information
Affiliations
Department of Anatomy, University of Otago, Dunedin, New Zealand
Neil J. Gemmell, Kim Rutherford, Tim A. Hore, Nicolas Dussex, Helen Taylor, Hideaki Abe & Donna M. Bond
LOEWE-Center for Translational Biodiversity Genomics, Senckenberg Museum, Frankfurt, Germany
Stefan Prost
South African National Biodiversity Institute, National Zoological Garden, Pretoria, South Africa
Stefan Prost
School of Life Sciences, Arizona State University, Tempe, AZ, USA
Marc Tollis, Melissa Wilson & Shawn M. Rupp
School of Informatics, Computing, and Cyber Systems, Northern Arizona University, Flagstaff, AZ, USA
Marc Tollis
School of Fundamental Sciences, Massey University, Palmerston North, New Zealand
David Winter
Peralta Genomics Institute, Oakland, CA, USA
J. Robert Macey, Charles G. Barbieri & Dustin P. DeMeo
School of Biological Sciences, The University of Adelaide, Adelaide, South Australia, Australia
David L. Adelson, Terry Bertozzi, Lu Zeng, R. Daniel Kortschak & Joy M. Raison
Department
of Ecology and Genetics – Evolutionary Biology, Evolutionary Biology
Centre (EBC), Uppsala University, Uppsala, Sweden
Alexander Suh, Valentina Peona, Claire R. Peart & Vera M. Warmuth
Department of Organismal Biology – Systematic Biology, Evolutionary Biology Centre (EBC), Uppsala University, Uppsala, Sweden
Alexander Suh & Valentina Peona
Evolutionary Biology Unit, South Australian Museum, Adelaide, South Australia, Australia
Nenhum comentário:
Postar um comentário
Observação: somente um membro deste blog pode postar um comentário.