Mitochondrial DNA can be inherited from fathers, not just mothers
A tenet of elementary biology is that mitochondria —
the cell’s powerhouses — and their DNA are inherited exclusively from
mothers. A provocative study suggests that fathers also occasionally
contribute.
The DNA of eukaryotic organisms (such as animals,
plants and fungi) is stored in two cellular compartments: in the
nucleus and in organelles called mitochondria, which transform nutrients
into energy to allow the cell to function. The nucleus harbours most of
our genes, tightly packaged into 46 chromosomes, of which half are
inherited from our mother’s egg and half from our father’s sperm. By
contrast, mitochondrial DNA (mtDNA) was thought to derive exclusively
from maternal egg cells, with no paternal contribution1. Writing in Proceedings of the National Academy of Sciences, Luo et al.2
challenge the dogma of strict maternal mtDNA inheritance in humans, and
provide compelling evidence that, in rare cases, the father might pass
on his mtDNA to the offspring, after all.
Human eggs contain more than 100,000 copies of mtDNA, whereas sperm contain approximately 100 copies3.
Early hypotheses suggested that paternal mtDNA molecules became diluted
in number relative to maternal mtDNA ones in the fertilized egg, but
these ideas were replaced when evidence from various organisms, such as
the uni-cellular alga Chlamydomonas reinhardtii4 and medaka fish5,
showed that paternal mtDNA is rapidly eliminated after fertilization.
For decades, researchers have speculated on why healthy organisms obtain
their cellular powerhouses from just one parent and on the possible
evolutionary advantages conferred by mitochondrial genes inherited in
this fashion.
A healthy individual’s mtDNA molecules are mostly
identical. But in people with diseases caused by mtDNA mutations, normal
and mutant mtDNA molecules typically coexist in a single cell — a
situation termed heteroplasmy6.
Disease severity is often associated with the amount of mutant mtDNA in
cells, which is in turn determined by events that occurred when the
person’s mother was still an embryo7.
The developing eggs in the female embryo go through an ‘mtDNA
bottle-neck’, in which the number of mtDNA copies is first reduced and
then amplified to more than 100,000 copies8,9.
Accordingly, variable amounts of mutant and normal mtDNA are present in
the mature eggs of an individual woman, and, therefore, in the cells of
her offspring. This phenomenon influences the severity of diseases
caused by mtDNA mutations, and can lead to very different manifestations
between individuals from the same family7.
Luo
and colleagues identified three families with mtDNA heteroplasmy that
could not be explained by maternal inheritance. The story started with a
young boy suspected of having a mitochondrial disease. The authors
performed high-resolution mtDNA sequencing, but did not identify any
disease-causing mtDNA mutations. However, their analysis uncovered
unusually high levels of mtDNA heteroplasmy. Intriguingly, the same
unusual pattern of mtDNA variation was found in the boy’s mother and in
his two healthy sisters (Fig. 1).
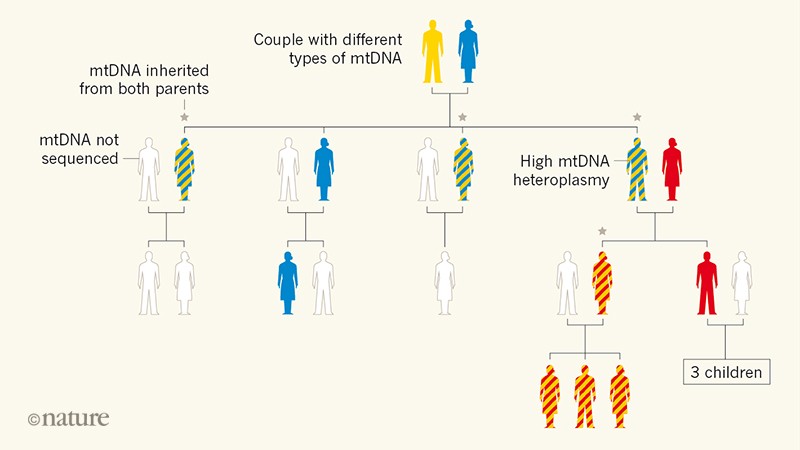
Figure 1 | Family tree revealing paternal inheritance of mitochondrial DNA (mtDNA). Luo et al.2
sequenced the mtDNA of several members of a family in which many
individuals had a high level of mtDNA heteroplasmy (the presence of
distinct genetic variants in the same cell). This mtDNA variability is
denoted by two colours in the same silhouette of an individual. The
analysis showed that some of the individuals with heteroplasmy had
inherited mtDNA from both of their parents, breaking the usual pattern
of exclusive maternal inheritance of mtDNA. Luo et al. suggest that the ability to inherit paternal mtDNA is a genetic trait.
To trace the origin of this mysterious mtDNA pattern, Luo et al.
extended their investigation to the previous generation. Sequencing of
the mtDNA of the boy’s maternal grandparents revealed an unexpected
contribution: his unusual mtDNA pattern seemed to be the product of
mtDNA from both grandparents. The authors went on to identify two
additional and unrelated families that had biparental mitochondrial
transmission. A similar scenario was previously observed in an
individual with mitochondrial disease who had a paternally inherited
mtDNA variant10. Together, these reports provide evidence for biparental mitochondrial inheritance in humans.
Human disease-causing mtDNA mutations were originally reported in 1988 (refs 6, 11)6,11, and more than 200 such mutations (see go.nature.com/2fucdqt) have been discovered since then, most of them occurring in a hetero-plasmic context7.
More-over, the estimated frequency of mutations of matrilineal mtDNA
has made it a useful and often-used tool in studies of ancestry and
evolution, as well as in forensic identification12.
Human mtDNA has also been a valuable tool in archaeology, because its
small size (16,569 base pairs) and circular form make it more resistant
to degradation than is nuclear DNA (which has around 3 billion base
pairs)13.
Given this long and multifaceted research history, why would paternal mtDNA have remained undetected? Luo et al.
suggest that mtDNA heteroplasmy is often overlooked in diagnostics when
it does not involve a disease-causing variant. Although this might be
true to some extent, it is a rather unsatisfactory explanation in this
era of deep DNA sequencing. Nevertheless, Luo and colleagues’ findings
should provoke a re-assessment of the extensive global mtDNA sequencing
data available, for those wishing to unearth further instances of
atypical heteroplasmy. If the paternal contribution to mtDNA is more
common than previously realized, this could alter some estimated timings
of human evolution, because these are often based on predictions of
mtDNA sequence variation under the assumption of exclusive maternal
inheritance.
Although biparental inheritance of mtDNA and
heteroplasmy coincided with disease symptoms in some of the individuals
studied by Luo et al., the authors’ data do not demonstrate a
causal link with disease. In fact, we cannot be certain that the study
participants have mitochondrial disease, because no specific
examinations to confirm this diagnosis are reported. Further study is
needed to identify more cases of potential paternal mtDNA inheritance,
and to determine the functional consequences of such heteroplasmy.
Notably, this knowledge is relevant to mitochondrial-donation therapy
(“three-parent babies”), which aims to prevent the transmission of
disease-causing mtDNA to offspring14, but which can also potentially generate individuals with two types of mtDNA, one from the donor and another from the mother.
Could
the amount of paternal mtDNA in a fertilized egg or developing embryo
be deliberately boosted to diminish the adverse effects of mutant
maternal mtDNA when this is present? This is an interesting option, but
still far from reality. In addition to evading elimination, paternal
mtDNA molecules would need to have a considerable replicative advantage
over maternal ones to reach meaningful proportions.
Will Luo and
colleagues’ findings affect the counselling of individuals carrying
disease-causing mtDNA mutations who are considering having children? Not
greatly, because paternal mitochondrial transmission seems to be
exceedingly rare in humans. At present, this discovery represents an
interesting conceptual breakthrough, rather than one that will directly
influence clinical practice.
Previous work15
has shown that mitophagy, the process by which cells ‘eat’ their own
mitochondria, has a role in the selective elimination of paternal
mitochondria. Given our rapidly expanding knowledge of mammalian
mitophagy in vivo16,
these rare instances of paternal mtDNA transmission might be attributed
to defective mitochondrial turnover.
The inheritance pattern of
paternal mtDNA in Luo and colleagues’ study suggests that a yet
unidentified gene on one of the autosomes (non-sex chromosomes) is
involved in eliminating paternal mitochondria. The families in whom
paternal mtDNA inheritance was observed provide an exciting opportunity
to decipher the signalling pathways that modulate paternal mitochondrial
elimination and prevent biparental mitochondrial transfer.

Nenhum comentário:
Postar um comentário
Observação: somente um membro deste blog pode postar um comentário.