A fonte da Peste Negra na Eurásia Central do século XIV
Natureza ( 2022 )
Abstrato
A origem da pandemia medieval de Peste Negra ( 1346-1353 ) tem sido um tópico de investigação contínua devido ao extenso impacto demográfico da pandemia e suas consequências duradouras 1 , 2 . Até agora, a evidência arqueológica mais debatida potencialmente associada ao início da pandemia deriva de cemitérios localizados perto do Lago Issyk-Kul do atual Quirguistão 1 , 3 , 4 , 5 , 6 , 7 , 8 , 9. Acredita-se que esses locais tenham abrigado vítimas de uma epidemia do século XIV, pois inscrições em lápides datadas diretamente de 1338-1339 indicam 'pestilência' como a causa da morte dos indivíduos enterrados 9 . Aqui relatamos dados de DNA antigos de sete indivíduos exumados de dois desses cemitérios, Kara-Djigach e Burana. Nossa síntese de dados arqueológicos, históricos e genômicos antigos mostra um claro envolvimento da bactéria da praga Yersinia pestis neste evento epidêmico. Dois antigos reconstruídos Y. pestisos genomas representam uma única cepa e são identificados como o ancestral comum mais recente de uma grande diversificação comumente associada ao surgimento da pandemia, aqui datada da primeira metade do século XIV. Comparações com a diversidade atual de reservatórios de Y. pestis na região estendida de Tian Shan suportam um surgimento local da cepa antiga recuperada. Por meio de várias linhas de evidência, nossos dados apoiam uma fonte do início do século XIV da segunda pandemia de peste na Eurásia central.
Principal
A Peste Negra, causada pela bactéria Y. pestis 10 , foi a onda inicial de uma pandemia de quase 500 anos denominada segunda pandemia de peste e é uma das maiores catástrofes de doenças infecciosas da história da humanidade 1 , 11 , 12 . Estima-se que tenha ceifado a vida de até 60% da população da Eurásia ocidental ao longo de seus oito anos 1 , 12 , a Peste Negra teve um profundo impacto demográfico e socioeconômico em todas as áreas afetadas, sendo o registro histórico europeu o mais extensamente recurso estudado até agora 2 , 13 , 14 , 15 .
Apesar da intensa pesquisa multidisciplinar sobre este tema, a origem geográfica da segunda pandemia de peste permanece incerta. Hipóteses baseadas em registros históricos e dados genômicos modernos apresentaram uma série de locais de origem putativos que variam da Eurásia Ocidental ao leste da Ásia (Informações Complementares 1 ). Nos últimos anos, comparações entre genomas de Y. pestis antigos e modernos mostraram que a Peste Negra está associada a uma emergência estrelada de quatro linhagens principais (ramos 1, 2, 3 e 4) 16 , 17 , cujos descendentes são dispersos entre focos de roedores na Eurásia, África e Américas. Embora linhagens existentes que divergiram antes deste evento tenham sido identificadas na Eurásia central e oriental 16, 18 , 19 , faltam dados complementares de DNA antigo (aDNA) dessas regiões. Até agora, as análises do registro histórico e dos dados antigos de Y. pestis se concentraram amplamente na progressão da pandemia no oeste da Eurásia 12 , 17 , 20 , 21 . Embora esforços para ampliar as investigações históricas e fornecer uma perspectiva espaço-temporal mais ampla estejam em andamento 9 , 11 , 22 , 23 , 24 , 25 , 26 , o foco eurocêntrico predominante tem dificultado a identificação das origens da Segunda Pandemia.
Uma epidemia do século XIV na Eurásia central
Para explorar possíveis evidências associadas à história inicial da segunda pandemia de peste, investigamos os cemitérios de Kara-Djigach e Burana, localizados no vale de Chüy, perto do lago Issyk-Kul, no atual Quirguistão. As escavações destes cemitérios entre 1885 e 1892 revelaram um conjunto arqueológico único potencialmente associado a uma epidemia que afectou a região durante o século XIV (Fig. 1 e Informação Complementar 2 ). Com base nas inscrições em lápides, esses cemitérios apresentaram um número desproporcionalmente alto de sepultamentos datados entre 1338 e 1339, com algumas inscrições afirmando que a causa da morte foi devido a uma pestilência não especificada 9 , 27 (Fig. 1) ., Dados Estendidos Fig. 1 , Fig. 1 Suplementar, Tabela 1 Suplementar e Informações Suplementares 2 ). Dada a localização, o momento e o padrão demográfico associado, as primeiras interpretações consideraram essas características como indicativas de uma epidemia de peste 3 , 27 e, desde então, desencadearam um longo debate sobre a associação da epidemia com o início da segunda pandemia de peste 1 , 3 , 4 , 5 , 6 , 7 , 8 , 9 , 26 (Informações Complementares 2).
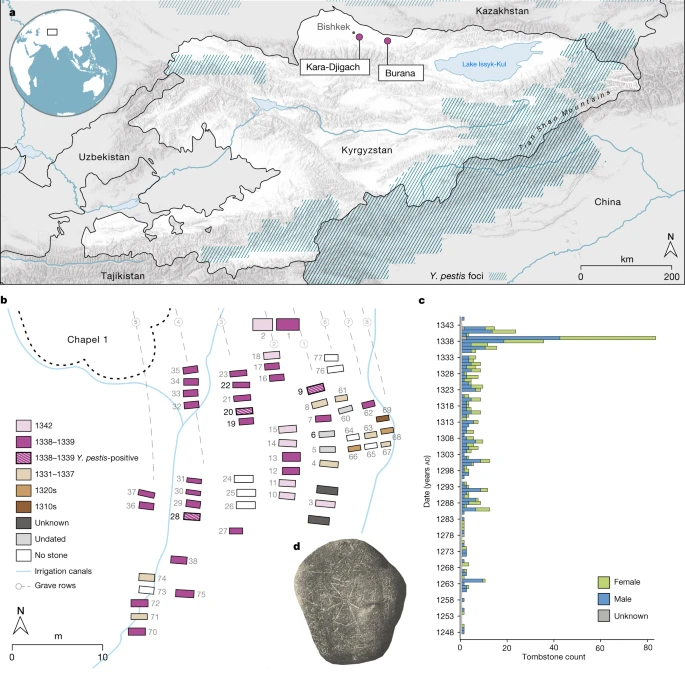
a , Localização dos sítios arqueológicos Kara-Djigach e Burana no atual Quirguistão. As regiões que abrangem os focos de Y. pestis atualmente estão destacadas em azul (como nas refs. 18 , 19 ). O mapa foi criado usando QGIS v.3.22.1 (ref. 51 ) e usa dados do mapa vetorial Natural Earth de https://www.naturalearthdata.com/ . b , Área dentro do cemitério Kara-Djigach, referida como 'Capela 1' com a maior concentração de sepulturas escavadas datadas entre 1338 e 1339. As datas de enterro foram determinadas com base nas lápides associadas (Informações Complementares 2). O mapa do local foi redesenhado com base no original criado por N. Pantusov em 1885. Indivíduos das sepulturas 6, 9, 20, 22 e 28 (os números em negrito) foram investigados usando DNA neste estudo. Enterros mostrados com padrões de listras foram associados aos indivíduos BSK001, BSK003 e BSK007, que mostraram evidências de infecções por Y. pestis . c , Números anuais de lápides de Kara-Djigach ( n = 456) e Burana ( n = 11) (Tabela Complementar 1 ). Conjunto de dados atualizado da ref. 9 (consulte Informações Complementares 2 para obter detalhes). d, Lápide do cemitério Kara-Djigach com inscrição legível associada à pestilência. A inscrição é traduzida como 'No ano de 1649 [= AD 1338], e foi o Ano do Tigre, em Turkic Bars. Este é o túmulo do crente Sanmaq. [Ele] morreu de peste [=mawtānā]'. Para um rastreamento da inscrição, consulte Dados Estendidos Fig. 1 .
Para entender melhor os contextos de Kara-Djigach e Burana, traduzimos e analisamos as informações de arquivo sobreviventes de suas escavações (Informações Suplementares 2 e Figs. 1 – 4 Suplementares ). Além disso, geramos dados genômicos humanos de 7 indivíduos (5 de Kara-Djigach e 2 de Burana) por meio de uma captura de hibridização de aproximadamente 1,24 milhão de polimorfismos de nucleotídeo único (SNPs) informativos de ancestralidade 28, o que resultou em 4 indivíduos com cobertura genômica suficiente para análises genéticas populacionais (>30.000 SNPs). Usando análise de componentes principais e modelagem de ancestralidade, descobrimos que esses indivíduos se enquadram amplamente na variabilidade de populações antigas e atuais da Eurásia central. No entanto, conexões precisas não puderam ser determinadas devido à escassez de dados genômicos humanos contemporâneos desta região (Informações Complementares 3 , Fig. 5 Complementar e Tabelas Complementares 2 - 5). Com base nas inscrições tumulares disponíveis, artefatos funerários, tesouros de moedas e registros históricos, descobrimos que o vale de Chüy abrigava comunidades etnicamente diversas que dependiam do comércio e mantinham conexões com várias regiões da Eurásia (Informações Complementares 2 ). Tais ligações podem ter contribuído para a disseminação de doenças infecciosas de e para esta região durante o século XIV.
Triagem de DNA de patógenos antigos
Para investigar vestígios de DNA de patógenos antigos que poderiam explicar a causa da suspeita de epidemia, dados metagenômicos de espingarda gerados de todos os sete indivíduos foram classificados taxonomicamente usando o pipeline HOPS 29 (Tabela Suplementar 6 ). Destes, três indivíduos exumados do cemitério Kara-Djigach (BSK001, BSK003 e BSK007) apresentaram evidências potenciais de DNA antigo de Y. pestis (Tabela Suplementar 7 ), bem como distâncias de edição baixas no mapeamento de leituras contra o genoma de referência CO92 e o presença de alterações químicas características do aDNA (Fig. 6 Complementar e Tabela 8 Complementar ). Como tal, as respectivas bibliotecas de DNA foram submetidas ao genoma completoCaptura de Y. pestis ( Métodos ).
O ancestral de uma politomia do século XIV
A captura do genoma inteiro de Y. pestis rendeu 6,7 vezes e 2,8 vezes a cobertura média para BSK001 e BSK003, respectivamente. A cobertura em todos os três plasmídeos de Y. pestis variou de 24,7 vezes a 4,7 vezes (Tabelas Suplementares 9 e 10 ). Para BSK007, a cobertura genômica foi menor, aproximadamente 0,13 vezes, resultante de uma preservação de aDNA mais pobre que também se refletiu na triagem de shotgun e nos dados de enriquecimento de DNA humano (Tabelas Suplementares 2, 3 e 8 ). No entanto, esta amostra foi considerada uma verdadeira Y. pestis - positiva devido à distribuição uniforme de leituras de mapeamento contra o cromossomo de referência CO92 e a presença de danos associados ao DNA (Figs.2 e 3 e Tabelas Suplementares 9 – 11 ). Além disso, uma classificação metagenômica de BSK007 lê o alinhamento com os plasmídeos pCD1, pMT1 e pPCP1 identificados >99% como específicos de Y. pestis (Dados Estendidos Fig. 3 ).
Para avaliar se os genomas de Y. pestis de maior cobertura BSK001 e BSK003 representavam cepas bacterianas distintas, comparamos seus perfis de SNP. Para limitar as chamadas variantes decorrentes da contaminação ambiental, particularmente devido às altas quantidades de sítios multi-alélicos identificados em ambos os genomas (Fig. 7 Suplementar ), realizamos uma filtragem metagenômica informada por taxonomia usando MALT ( Métodos e Tabela Suplementar 11 ). Identificamos 20 locais que diferem entre BSK001 e BSK003, todos os quais são variantes únicas no BSK003 de cobertura inferior (Tabela Suplementar 12 ). Com base em critérios de autenticidade previamente definidos 30 , 31 ( Métodos), todas essas variantes eram consistentes com contaminação exógena residual, sugerindo que os dois genomas eram provavelmente idênticos. A recuperação de cepas idênticas de ambos os indivíduos é consistente com evidências publicadas mostrando baixa diversidade em genomas de Y. pestis isolados de contextos epidêmicos únicos 10 , 17 , 20 , 21 , 32 . Com base em suas lápides associadas, BSK001, BSK003 e BSK007 foram enterrados durante o ano epidêmico 1338-1339 (Fig. 1 e Informações Suplementares 2 ) e nossos dados apoiam ainda mais o envolvimento de Y. pestis neste evento.
Realizamos uma análise comparativa de SNP entre os genomas de Kara-Djigach e a diversidade histórica e circulante de Y. pestis previamente publicada (Fig. 2a , Tabelas Suplementares 13 – 15 ). Para isso, BSK001 e BSK003 foram combinados (BSK001/003) para obter uma resolução genômica aumentada (cobertura combinada de 9,5 vezes; Tabela Suplementar 9 ). Nossa análise revelou um SNP exclusivo para BSK001/003 quando comparado com 203 genomas cromossômicos modernos e 46 históricos de Y. pestis (Dados Estendidos Fig. 4 e Tabelas Suplementares 16 e 17). Este SNP foi encontrado em uma região com sítios multialélicos persistentes; portanto, é considerado artefactual 31 (Fig. 8 Complementar ). Consistente com pesquisas anteriores sobre a história evolutiva de Y. pestis 16 , nossa filogenia de máxima verossimilhança inferida exibiu cinco ramos principais, designados 0, 1, 2, 3 e 4, com genomas publicados da Segunda Pandemia sendo associados ao ramo 1 (Fig. 2b ) . A colocação de BSK001/003 é ancestral de todos os genomas publicados do século XIV da Eurásia ocidental (Fig. 2b e Dados Estendidos Fig. 5 ), separados por um SNP de LAI009, um isolado da região do Volga na Europa Oriental 17, e por dois SNPs de cinco genomas geneticamente idênticos associados à Morte Negra do sul, centro e norte da Europa 17 , 21 . Especificamente, BSK001/003 está posicionado em um nó previamente designado N07 (ref. 16 ), que precedeu a multifurcação dos ramos 1–4. Para avaliar se os dados ausentes afetaram a precisão de nossos posicionamentos filogenéticos, investigamos todas as chamadas variantes BSK001 e BSK003 para posições compartilhadas com linhagens derivadas do nó N07 e aquelas que o precedem diretamente. BSK001/003 carrega o estado ancestral em todos os SNPs diagnósticos cobertos que definem os ramos 1-4 e 0.ANT3, que é a linhagem do ramo 0 mais próxima de BSK001/003, bem como o estado derivado em todas as posições que vão de 0.ANT3 a N07 (Fig. 2c, Dados Estendidos Fig. 6 e Tabela Suplementar 18 ). Em nossa resolução atual, concluímos que BSK001/003 representa o progenitor direto da politomia do ramo 1-4.
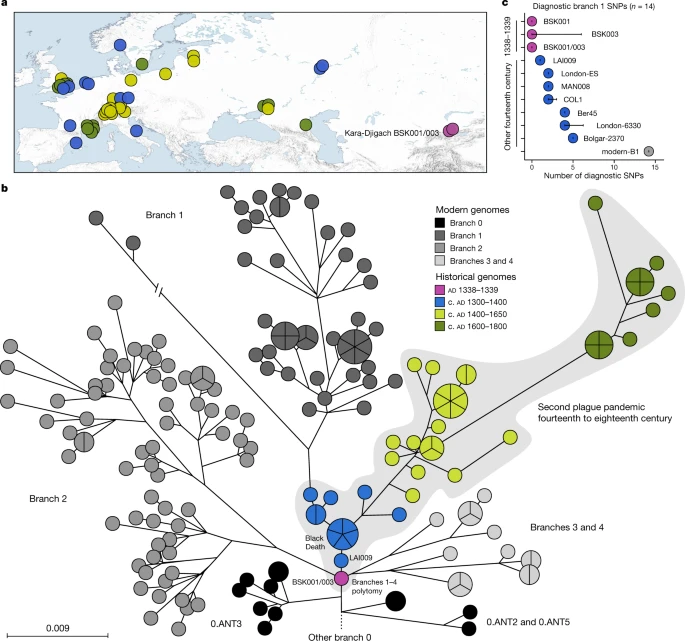
a , Mapa de todos os genomas históricos de Y. pestis usados no presente estudo ( n = 48). As cores representam diferentes idades do genoma em uma escala entre 1300 e 1800, conforme ilustrado em b . A escala de cores é mantida em todos os painéis desta figura. Para ajudar a visibilidade em símbolos sobrepostos, uma opção de jitter foi implementada para plotar genomas no mapa. O mapa foi criado com QGIS v.3.22.1 (ref. 51 ) e usa dados do mapa vetorial Natural Earth de https://www.naturalearthdata.com/ . b , filogenia de máxima verossimilhança de Y. pestis com base em 2.960 SNPs, visualizados usando GrapeTree 50. A porção representada da filogenia contém as linhagens relacionadas mais próximas de BSK001/003. (Para uma árvore totalmente rotulada, veja Dados Estendidos Fig. 5 ). As cores das linhagens históricas publicadas são consistentes com um . A escala denota o número de substituições por sítio genômico. c , Abundância de compartilhamento de SNP diagnóstico em genomas de Y. pestis do século XIV . O número de SNPs diagnósticos ( n ) compartilhados entre todos os genomas modernos no ramo 1 e, portanto, definindo esse ramo, foram recuperados de uma tabela comparativa de SNPs de 203 Y. pestis modernosgenomas. O compartilhamento de SNP foi avaliado determinando o status do alelo de cada posição diagnóstica de acordo com um limiar de chamada de SNP triplo. As barras de erro denotam o grau de falta de dados ( n ) no respectivo genoma antigo. Consulte a Fig. 6 de Dados Estendidos e a Tabela Suplementar 18 para obter uma visão geral do compartilhamento de SNP de diagnóstico em diferentes ramos filogenéticos.
Tempo de divergência para a politomia do ramo 1-4
A politomia dos ramos 1-4 é um evento importante na evolução de Y. pestis , dada a sua associação com a Peste Negra 9 , 26 , 33 e a rica diversidade genética que dela emergiu 16 (Fig. 2b ). As estimativas sobre o momento dessa diversificação até agora renderam amplas variações que vão desde os séculos X ao XIV 16 , 34 . Recentemente, foi proposto um recorte temporal mais estreito que situava esse surgimento no início do século XIII, mais de 100 anos antes da Peste Negra 22 , 26. Como BSK001/003 representa o ancestral comum dos ramos 1-4, usamos esse genoma de 1338 a 1339 para construir uma filogenia calibrada no tempo e reestimar uma faixa etária para essa diversificação com BEAST2 (Figs . Tabela 19 ). Depois de avaliar uma série de modelos demográficos (Tabela Suplementar 20 ), nossas estimativas resultantes com base no modelo de horizonte Bayesiano coalescente revelaram idades sobrepostas para a divergência de BSK001/003 (95% maior densidade posterior (HPD): 1308-1339), bem como quanto ao ramo 1 dos ramos 2–4 (95% HPD: 1317–1345) (Fig. 3 ). Como o BEAST2 infere apenas árvores bifurcadas, também usamos TreeTime 35para inferir uma filogenia calibrada no tempo que pode reter politomias. Consistente com nossas estimativas acima, inferimos uma data de 1316-1340 para o tempo de divisão dos ramos 1-4 (Fig. 11 suplementar ), embora advertimos que esse método não leva em conta incertezas de idade em genomas antigos. Tomados em conjunto, os presentes resultados suportam uma faixa etária que abrange a primeira metade do século XIV para o momento da politomia do ramo 1-4.
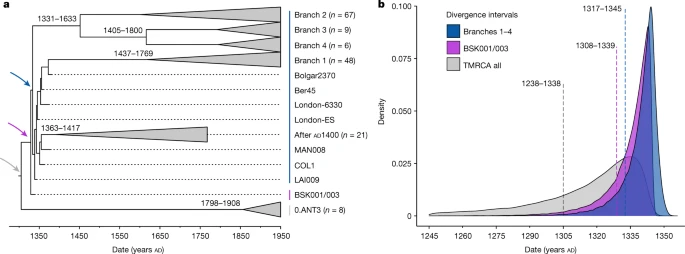
a , Árvore filogenética calibrada no tempo de credibilidade máxima do clado. A árvore é baseada em 167 genomas (históricos e modernos) e foi estimada usando a árvore do horizonte coalescente anterior e um relógio relaxado log-normal. Galhos caídos contêm isolados modernos e antigos que datam de 1400 DC (pós-Peste Negra). As setas coloridas marcam os nós, para os quais as distribuições de idade posteriores equivalentes são mostradas em b . As datas de divergência estimadas (intervalos HPD de 95%) das ramificações modernas são mostradas em cada nó correspondente. b , Distribuições posteriores estimadas com base na árvore do horizonte coalescente Bayesiana anterior para a divergência de Y. pestisramos 1–4 (azul), para a divergência estimada de BSK001/003 (roxo) e para todo o conjunto de dados usado para esta análise (tempo para o ancestral comum mais recente dos ramos 1–4 e 0.ANT3, mostrado em cinza) . As linhas pontilhadas indicam estimativas posteriores médias e são anotadas com os correspondentes intervalos HPD de 95%.
Além disso, para quantificar a proporção da diversidade genética atual de Y. pestis que emergiu dessa politomia, calculamos as distâncias médias aos pares (MPDs) e os índices de diversidade filogenética de Faith (FPD) em 203 genomas que compõem todo o nosso conjunto de dados moderno, bem como 130 genomas que compreendem os ramos 1–4 ( Métodos ). Em nosso conjunto de dados, 64% (130 de 203) das cepas modernas de Y. pestis pertenciam aos ramos 1-4, refletindo a alta frequência mundial conhecida dessas linhagens 16 , 36 , 37 . Estimamos que os ramos 1-4 representam aproximadamente 40% da diversidade filogenética geral dentro da atual Y. pestiscom base em nosso conjunto de dados completo (proporção MPD: 41%; intervalo de percentil de 95% (PI): 35,3–46,4; proporção FPD: 35,9%; 95% PI: 31,6–39,5). Este valor é marginalmente reduzido após equalizar o número de genomas nos ramos 1–4 e ramo 0 (proporção MPD: 36,8%; 95% PI: 32,0–41,9; proporção FPD: 33,9%; 95% PI: 29,4–37,7) (Extended Dados Fig. 7 ). Dado que a história conhecida de Y. pestis remonta a pelo menos 5.000 anos 38 , é notável que uma fração substancial de sua diversidade genética sobrevivente se acumulou desde o século XIV.
Reservatórios de peste na área de Tian Shan
Para abordar as hipóteses existentes sobre as origens geográficas da Peste Negra (Informações Complementares 1 ), investigamos a possibilidade de uma emergência local versus uma introdução da cepa BSK001/003 no Vale do Chüy de uma área diferente. Para isso, avaliamos a distribuição geográfica das linhagens ramificadas ancestrais mais estreitamente relacionadas com BSK001/003 e identificamos 164 cepas 0.ANT atuais com registro de seus locais de isolamento (Tabela Complementar 21 ). Consistente com interpretações anteriores 9 , 18 , 26 , descobrimos que todas essas cepas foram recuperadas de focos no leste do Cazaquistão, leste do Quirguistão e da Região Autônoma Uigur de Xinjiang no noroeste da China (Fig. 4e Dados Estendidos Fig. 8 ). Embora não possamos excluir uma faixa geográfica diferente para essas linhagens no passado, os dados atuais são consistentes com o surgimento local de BSK001/003 na região estendida de Tian Shan. Curiosamente, o genoma mais antigo recuperado associado ao 0.ANT também foi identificado na região de Tian Shan (século III DC ) 39 e faz parte de um clado extinto que causou a primeira pandemia de peste (séculos VI ao VIII DC ) 30 . Como observado anteriormente 18 , 26 , 33 , 40 , a maioria das cepas 0.ANT existentes foram isoladas de marmotas e seus ectoparasitas conhecidos por serem os principaisreservatórios de Y. pestis nessas áreas (Tabela Complementar 21 ). Portanto, tais espécies podem representar possíveis candidatos ao transbordamento que levou à segunda pandemia de peste.
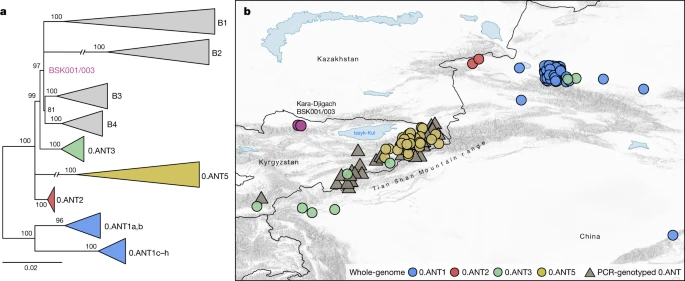
a , Árvore filogenética de máxima verossimilhança, com base em 2.441 posições variantes do genoma. A árvore foi construída para indicar as relações genéticas entre os genomas 0.ANT disponíveis representados no mapa e BSK001/003. Os ramos modernos foram recolhidos para aumentar a clareza da árvore (veja Dados Estendidos Fig. 8 para uma árvore completa). b , Mapa representando os locais de isolamento geográfico de cepas 0.ANT (Tabela Suplementar 21), que pertencem às linhagens ramificadas ancestrais mais próximas da linhagem Kara-Djigach. O mapa inclui dados de genoma completo (especificados como 0.ANT linhagens 1, 2, 3 e 5) e isolados genotipados por PCR que são amplamente definidos como 0.ANT, pertencentes a qualquer uma das 4 linhagens. Para as cepas em que as coordenadas geográficas exatas não estavam disponíveis, as localizações foram aproximadas de acordo com seus reservatórios de peste associados. Para auxiliar a visibilidade em símbolos sobrepostos, uma opção de jitter foi implementada para plotar objetos no mapa. O mapa foi criado com QGIS v.3.22.1 (ref. 51 ) e usa dados do mapa vetorial Natural Earth de https://www.naturalearthdata.com/ .
Discussão
O poder da metagenômica antiga reside em seu potencial de fornecer evidências diretas para testar hipóteses históricas de longa data e revelar padrões filogeográficos de diversidade microbiana ao longo do tempo 41 . Um desses debates diz respeito aos eventos que desencadearam a segunda pandemia de peste, bem como a hora e o local de seu surgimento. Recentemente, uma análise de dados históricos, genéticos e ecológicos levou à sugestão de que o surgimento dos ramos 1-4 de Y. pestis ocorreu mais de um século antes do início da Peste Negra. De acordo com o modelo proposto, essa diversificação inicial foi mediada por pessoas e estava ligada às expansões territoriais do Império Mongol pela Eurásia durante o início do século XIII 22 , 26 ,42 . Em contraste, apresentamos dados antigos de Y. pestis da Eurásia central que apóiam um surgimento no século XIV; portanto, as atribuições de surtos anteriores ainda precisam ser exploradas. Atualmente, a amostragem de foco estreito escolhida para este estudo não permite uma avaliação da disseminação da cepa BSK001/003. Estudos anteriores mostraram que Y. pestis pode se disseminar rapidamente sem acúmulo de diversidade genética 17 , 21, potenciando assim a presença contemporânea da mesma estirpe numa vasta área geográfica. No entanto, a gama conhecida de focos de peste existentes associados a linhagens ancestrais de BSK001/003 fornece suporte para seu surgimento na Eurásia central e possivelmente na região estendida de Tian Shan. Embora a dinâmica que desencadeou o surgimento da bactéria nesta região seja desconhecida, estudos anteriores mostraram que fatores ambientais, como desastres naturais e mudanças bruscas de temperatura e precipitação podem ter impacto nas ecologias de hospedeiros de Y. pestis e, consequentemente, podem desencadear surtos em populações humanas 43 , 44 , 45 , 46. Embora não tenhamos evidências para inferir tais conexões com a epidemia de Kara-Djigach, prevemos que nossa data precisa de 1338-1339 servirá como ponto de referência para futuras pesquisas ambientais, arqueológicas e históricas com foco nos eventos que causaram a introdução de Y. pestis em populações humanas e precipitou a segunda pandemia de peste.
O início da Peste Negra tem sido convencionalmente associado a surtos ocorridos na região do Mar Negro em 1346 (refs. 1 , 47 ), oito anos após a epidemia de Kara-Djigach. Atualmente, os meios exatos pelos quais Y. pestis atingiu a Eurásia ocidental são desconhecidos, principalmente devido a grandes incertezas pré-existentes em torno dos contextos históricos e ecológicos desse processo. Pesquisas anteriores sugeriram que as redes de guerra e/ou comércio foram alguns dos principais contribuintes na disseminação de Y. pestis 21 , 22 , 26 , 47 , 48. No entanto, estudos relacionados até agora se concentraram em expedições militares que não estavam relacionadas a surtos iniciais 47 ou outros que ocorreram muito antes de meados do século XIV 22 , 26 . Além disso, embora existam análises preliminares para apoiar um envolvimento das rotas comerciais da Eurásia na propagação da doença 48 , sua exploração sistemática até agora foi conduzida apenas para áreas restritas da Eurásia ocidental 21 , 47 . A colocação do assentamento Kara-Djigach nas proximidades de redes transasiáticas 9 , 49 , bem como as diversas evidências toponímicas e artefatos identificados no local (Informações Complementares 2 ) dão suporte a cenários que implicam o comércio na disseminação de Y. pestis . Portanto, uma investigação das conexões do início a meados do século XIV em toda a Ásia, interpretada ao lado de evidências genômicas, será importante para desvendar as dispersões da bactéria para o oeste.
Experiências passadas e presentes demonstraram que reconciliar a origem de uma pandemia é uma tarefa complexa que não pode ser realizada por uma única disciplina de pesquisa. Embora os genomas antigos de Y. pestis relatados neste artigo ofereçam evidências biológicas para resolver um antigo debate, são os contextos históricos e arqueológicos únicos que definem o escopo e a importância de nosso estudo. Como tal, vislumbramos que as sinergias futuras continuarão a revelar insights importantes para uma reconstrução detalhada dos processos que desencadearam a segunda pandemia de peste.
Métodos
Amostragem, extração de DNA, preparação e sequenciamento de biblioteca de glicosilase de DNA de uracila parcial
Obtivemos permissão do Kunstkamera, Peter the Great Museum of Anthropology and Ethnography em São Petersburgo para a amostragem e análise de DNA antigo de 7 espécimes de dentes, escavados entre 1885 e 1892 nos cemitérios medievais de Kara-Djigach e Burana (Informações Complementares 2 ) . Não foram utilizados métodos estatísticos para predeterminar o número de amostras utilizadas neste estudo. Todos os procedimentos laboratoriais foram realizados nas instalações dedicadas de aDNA do Instituto Max Planck para a Ciência da História Humana e do Instituto Max Planck para Antropologia Evolutiva. Os procedimentos detalhados usados para amostragem de dentes podem ser encontrados na ref. 52. Resumidamente, os dentes foram seccionados na junção dentina-esmalte usando uma serra elétrica com lâmina diamantada. Após o seccionamento do dente, aproximadamente 50 mg de pó foram removidos da superfície da câmara pulpar de cada dente usando brocas odontológicas arredondadas.
O pó do dente recuperado foi utilizado para extrações de DNA utilizando um protocolo previamente estabelecido otimizado para a recuperação de fragmentos curtos de DNA 53 . As etapas exatas e as modificações do procedimento utilizado foram disponibilizadas na ref. 54 . Em resumo, o pó dental foi incubado durante a noite (12–16 h) a 37°C em 1 ml de tampão de lise de DNA contendo EDTA (0,45 M, pH 8,0) e proteinase K (0,25 mg ml- 1). Após a incubação, a ligação e o isolamento do DNA foram realizados usando um tampão de ligação à base de GuHCl personalizado e a purificação usando o Kit de Grande Volume de Ácido Nucleico Viral de Alta Pureza (Roche). Finalmente, o DNA foi eluído em 100 μl de Tris-EDTA-Tween contendo Tris-HCl (10 mM), EDTA (1 mM, pH 8,0) e Tween-20 (0,05%). Para o monitoramento do procedimento, os brancos de extração e os controles de extração positivos foram incluídos em todas as etapas de processamento do laboratório.
Todos os extratos de DNA foram convertidos em uma a duas bibliotecas de DNA de fita dupla para sequenciamento Illumina, usando 25 μl de extrato de entrada por biblioteca com uma glicosilase de DNA de uracila parcial inicial (UDG) e tratamento com endonuclease VIII (enzima USER; New England Biolabs) conforme protocolos estabelecidos 55 , 56 . O procedimento detalhado de preparação da biblioteca, incluindo as etapas de reparo da extremidade cega, ligação do adaptador e reação de preenchimento do adaptador, podem ser encontrados na ref. 57 . Após a preparação da biblioteca, cada biblioteca foi quantificada usando um sistema de PCR quantitativo (LightCycler 96 Instrument) usando os primers IS7 e IS8 55 . Para sequenciamento multiplex, realizamos dupla indexação de todas as bibliotecas usando procedimentos publicados anteriormente 58, descrito em detalhes na ref. 59 . Uma combinação de primers de índice únicos contendo identificadores de 8 pares de bases (bp) foram atribuídos a cada biblioteca. Para ajudar na eficiência da amplificação, as bibliotecas foram então divididas em várias reações de PCR para a etapa de indexação com base em sua quantificação inicial de IS7/IS8. O número de reações de PCR de indexação realizadas para cada biblioteca foi determinado de modo que cada reação recebeu uma entrada de não mais que 1,5 × 10 10cópias de DNA. Cada reação foi configurada usando a polimerase de DNA Pfu Turbo Cx Hotstart (Agilent Technologies) e foi executada por 10 ciclos usando as seguintes condições: desnaturação inicial a 95 °C por 2 min seguido por um ciclo de 95 °C por 30 s, 58 °C por 30 s e 72 °C por 1 min, bem como uma etapa final de alongamento a 72 °C por 10 min. Todos os produtos de PCR foram purificados com o MinElute DNA Purification Kit (QIAGEN), com algumas modificações no protocolo do fabricante 59 . Finalmente, todos os produtos de PCR de indexação foram quantificados por qPCR (LightCycler 96 Instrument) usando a combinação de primers IS5 e IS6 58 , 59 . Para evitar a formação de heteroduplex, as bibliotecas indexadas foram amplificadas para 10 13Cópias de DNA por reação com a Herculase II Fusion DNA Polymerase (Agilent Technologies) e quantificadas usando um instrumento 4200 Agilent TapeStation usando um sistema D1000 ScreenTape (Agilent Technologies). As bibliotecas foram diluídas para 10 nM e reunidas equimolarmente para sequenciamento. Realizamos o sequenciamento de DNA shotgun em uma plataforma Illumina HiSeq 4000 usando um kit de 76 ciclos (1 × 76 + 8 + 8 ciclos).
Processamento de leitura de sequenciamento de próxima geração e triagem metagenômica Shotgun
Após a demultiplexação, as leituras sequenciadas de shotgun brutas foram pré-processadas no pipeline EAGER v.1.92.58 usando AdapterRemoval v.2.2.0 (ref. 60 ), que foi usado para remover adaptadores Illumina (sobreposição mínima de 1 bp), bem como para filtragem de leitura de acordo com a qualidade de sequenciamento (qualidade de base mínima de 20) e comprimento (retenção de leituras ≥30 bp). Posteriormente, todos os conjuntos de dados foram selecionados para a presença de traços de DNA de patógenos usando o pipeline metagenômico HOPS 29 . Primeiro, as leituras pré-processadas foram alinhadas com um banco de dados RefSeq personalizado 61 (novembro de 2017) contendo todos os conjuntos completos de genomas bacterianos e virais, um subconjunto de conjuntos de patógenos eucarióticos e o GRCh38genoma de referência humano. As montagens do genoma que continham a palavra 'desconhecido' foram removidas do banco de dados, mantendo um total de 15.361 entradas. O banco de dados reteve várias entradas de espécies de Yersinia : Yersinia aldovae ( n = 1), Yersinia aleksiciae ( n = 1), Yersinia enterocolitica ( n = 16), Yersinia entomophaga ( n = 1), Yersinia frederiksenii ( n = 3), Yersinia intermedia ( n = 1), Yersinia kristensenii ( n = 2), Y. pestis (n = 39), fago Yersinia ( n = 17), Yersinia pseudotuberculosis ( n = 13), Yersinia rohdei ( n = 1), Yersinia ruckeri ( n = 4), Yersinia similis ( n = 1) e Yersinia sp. FDA-ARGOS ( n = 1). MALT v0.4 62foi executado usando os seguintes parâmetros: -id 90 -lcaID 90 -m BlastN -at SemiGlobal -topMalt 1 -sup 1 -mq 100 -verboseMalt 1 -memoryMode load -additionalMaltParameters. Os arquivos de alinhamento resultantes foram pós-processados com MALTExtract para uma avaliação qualitativa em relação a uma lista predefinida de 356 entradas taxonômicas de destino ( https://github.com/rhuebler/HOPS/blob/external/Resources/default_list.txt ). Especificamente, as leituras foram avaliadas de acordo com sua distância de edição em relação a uma sequência de patógeno específica no banco de dados e a possível ocorrência de incompatibilidades que poderiam significar a presença de danos no DNA 29 . Nos casos em que ambos os parâmetros foram atendidos, o alinhamento do patógeno correspondente foi considerado um forte candidato. As leituras pré-processadas foram mapeadas contra a Y. pestisCO92 (NC_003143.1) e genomas de referência humanos ( hg19 ) com o alinhador Burrows-Wheeler (BWA). Os parâmetros de mapeamento foram definidos para 0,01 para a distância de edição (-n) e o comprimento da semente foi desabilitado (-l 9999). Posteriormente, usamos SAMtools v.1.3 (ref. 63 ) para remover reads com qualidade de mapeamento inferior a 37 (para CO92) ou 30 (para hg19 ); As duplicatas de PCR foram removidas com MarkDuplicates v1.140 ( http://broadinstitute.github.io/picard/ ). Finalmente, os padrões de dano de aDNA foram avaliados com mapDamage v.2.0 (ref. 64 ).
Preparação de biblioteca de DNA de fita simples e captura de hibridização
Para os espécimes BSK001 e BSK003, bibliotecas extras de DNA de fita simples foram construídas a partir de um extrato de DNA de entrada de 30 μl. Realizamos a preparação da biblioteca no Instituto Max Planck de Antropologia Evolutiva usando um protocolo automatizado que está disponível publicamente 65 . Bibliotecas de fita simples e fita dupla de indivíduos BSK001, BSK003 e BSK007 foram enriquecidas usando sondas de DNA cobrindo todo o genoma de Y. pestis , bem como 1,24 milhão de sítios SNP de todo o genoma do genoma humano 66 , 67. Para a preparação da captura, todas as bibliotecas foram amplificadas para o número necessário de ciclos de PCR para atingir 1–2 μg de DNA de entrada. As reações de PCR foram realizadas usando a Herculase II Fusion DNA Polymerase. Eles foram então purificados usando o MinElute DNA Purification Kit e eluídos em tampão de eluição EB contendo 0,05% de Tween 20. Finalmente, as concentrações da biblioteca (ng μl −1 ) foram quantificadas usando um espectrofotômetro NanoDrop (Thermo Fisher Scientific). Para as capturas de Y. pestis em solução , o projeto do conjunto de sondas foi baseado em um conjunto de genomas modernos disponíveis publicamente, especificamente o cromossomo Y. pestis CO92 (NC_003143.1), plasmídeo CO92 pMT1 (NC_003134.1), plasmídeo CO92 pCD1 (NC_003131.1), cromossomo KIM10 (NC_004088.1), cromossomo F Pestoides (NC_009381.1) e oCromossomo Y. pseudotuberculosis IP32953 (NC_006155.1). Para as capturas de DNA humano em solução, o design do conjunto de sondas foi criado para atingir 1.237.207 variantes em todo o genoma que são informativas para estudar a história genética de populações humanas em todo o mundo 28 , 67 . As capturas de hibridização de DNA humano e Y. pestis foram realizadas por duas rodadas, conforme descrito anteriormente 28 , 69 , 68 , 67 , 66 , em que bibliotecas parcialmente tratadas com UDG do mesmo indivíduo foram agrupadas em razões equimolares para captura e fita simples bibliotecas foram capturadas separadamente.
Processamento de dados pós-captura de Y. pestis
Após a captura do genoma completo de Y. pestis , as bibliotecas foram sequenciadas em uma plataforma HiSeq 4000 (1 × 76 + 8 + 8 ciclos ou 2 × 76 + 8 + 8 ciclos) em uma profundidade de aproximadamente 11–27 milhões de leituras brutas. O pré-processamento de leituras desmultiplexadas brutas foi realizado conforme descrito na seção 'Processamento de leitura de sequenciamento de próxima geração do Shotgun e triagem metagenômica'. Nesta fase, os conjuntos de dados produzidos a partir de bibliotecas parcialmente tratadas com UDG do mesmo indivíduo foram agrupados e as bases terminais foram cortadas usando fastx_trimmer (FASTX Toolkit 0.0.14, http://hannonlab.cshl.edu/fastx_toolkit/ ) para evitar danos ao site interferência com a chamada SNP durante o processamento posterior. As etapas a seguir para mapeamento de leitura, remoção de duplicatas de PCR e cálculo de danos de aDNA foram realizadas no pipeline EAGER70 . Realizamos mapeamento de leitura com BWA v.0.7.12 contra o genoma de referência de Y. pestis CO92 (NC_003143.1). Para as bibliotecas tratadas com UDG parcial agrupadas e aparadas, os parâmetros BWA foram definidos para 0,1 para a distância de edição (-n) e o comprimento da semente foi desativado (-l 9999). Dado que as bibliotecas de fita simples construídas para este estudo retiveram danos associados ao DNA, os parâmetros BWA foram definidos para 0,01 para a distância de edição (-n) para permitir um número maior de incompatibilidades que poderiam derivar da desaminação; o comprimento da semente foi desabilitado (-l 9999). Realizamos mapeamento de leitura contra os plasmídeos usando os mesmos parâmetros contra uma referência concatenada de todos os três Y. pestisplasmídeos (pMT1: NC_003134.1; pPCP1: NC_003132.1; e pCD1: NC_003131.1), mascarando a região pPCP1 problemática entre os nucleotídeos 3000 e 4200 que mostrou ter alta similaridade com vetores de expressão usados em reagentes de laboratório 71 . SAMtools v.1.3 (ref. 63 ) foi usado para remover todas as leituras com qualidade de mapeamento inferior a 37 (-q), enquanto MarkDuplicates foi usado para remover duplicatas de PCR. Padrões de desaminação associados com dano de aDNA foram recuperados com mapDamage v.2.0 (ref. 64 ). Usamos MALT 62 para uma classificação taxonômica de leituras mapeadas, para tentar uma retenção de leituras que são mais prováveis de serem endógenas de Y. pestis. O MALT foi executado no mesmo banco de dados descrito na seção 'Processamento de leitura de sequenciamento de próxima geração do Shotgun e triagem metagenômica', usando os seguintes parâmetros: -m BlastN -at SemiGlobal -top 1 -sup 1 -mq 100 -memoryMode load -ssc -sp. O parâmetro de identidade de porcentagem mínima foi definido como padrão (-id 0,0), em oposição a um filtro de identidade de 90% usado para executar o HOPS 29 , para evitar qualquer viés de referência que possa surgir da remoção de leituras endógenas com um número maior de incompatibilidades. Após a conclusão da execução, para manter o número máximo de leituras contabilizando o algoritmo ingênuo do ancestral comum mais baixo, extraímos as leituras que foram atribuídas ao nó do gênero Yersinia ou resumidas sob o Y. pseudotuberculosisnó complexo. As leituras foram extraídas no formato FASTA de MEGAN v.6.4.12 (ref. 72 ). Posteriormente, os arquivos FASTA foram convertidos para o formato FASTQ com o script reformat.sh no BBMap da suíte BBtools (versão 38.86, https://sourceforge.net/projects/bbmap/ ). Os arquivos FASTQ foram então remapeados contra o genoma de referência CO92 usando os mesmos parâmetros descritos anteriormente nesta seção. Para bibliotecas de fita simples, mapDamage v.2.0 (ref. 64) foi usado para redimensionar as pontuações de qualidade em posições de leitura nas quais foram identificadas possíveis incompatibilidades associadas à desaminação para a referência. Posteriormente, os arquivos BAM correspondentes ao mesmo indivíduo foram concatenados após filtragem de qualidade do mapeamento e remoção de duplicatas de PCR. Realizamos a concatenação usando o comando 'merge' do SAMtools e com a ferramenta AddOrReplaceReadGroups no Picard ( http://broadinstitute.github.io/picard/ ) para atribuir um único grupo de leitura a todas as leituras em cada novo arquivo.
Chamada de SNP, estimativas de heterozigosidade e filtragem de SNP
A chamada de variantes foi realizada para BSK001 e BSK003, antes e depois da filtragem MALT 62 usando o UnifiedGenotyper no Genome Analysis Toolkit (GATK) v.3.5 (ref. 73 ). O GATK foi executado usando a opção EMIT_ALL_SITES, que produziu uma chamada para cada posição no genoma de referência cromossômico CO92. Os perfis genômicos resultantes de BSK001 e BSK003 foram comparados com um conjunto de 233 genomas modernos e 46 históricos de Y. pestis , bem como com o genoma de referência de Y. pseudotuberculosis IP32953 (NC_006155.1), usando a ferramenta Java MultiVCFAnalyzer v.0.85 ( https://github.com/alexherbig/MultiVCFAnalyzer). O MultiVCFAnalyzer v.0.85 foi executado com os seguintes parâmetros. Os SNPs foram chamados com uma cobertura mínima de três vezes e em casos de posições heterozigóticas, as chamadas foram feitas com um limite mínimo de suporte de 90%. Além disso, SNPs foram chamados com uma qualidade mínima de genotipagem de 30. Além disso, regiões não-core e repetitivas previamente definidas, bem como regiões contendo homoplasias, RNAs ribossômicos, RNAs de transferência-mensageiro e RNAs de transferência foram excluídos da chamada SNP comparativa 16 , 32 . Um conjunto de 6.567 locais variantes totais foi identificado no presente conjunto de dados.
Para investigar a extensão da possível contaminação exógena nos conjuntos de dados BSK001 e BSK003, estimamos o número de variantes heterozigóticas ambíguas além do limite de chamada do SNP. Para isso, foi utilizado o MultiVCFAnalyzer v.0.85 (ref. 74 ) para gerar uma tabela SNP de frequências alélicas alternativas variando entre 10 e 90%. Os resultados foram então usados para criar gráficos de histograma de 'heterozigosidade' das frequências estimadas em R v.3.6.1 (ref. 75 ). Gráficos de heterozigosidade foram criados antes e depois da filtragem MALT (consulte ' Processamento de dados de Y. pestis pós-captura ') para investigar se a filtragem informada por taxonomia poderia ajudar na eliminação de sequências contaminantes nos conjuntos de dados investigados (Fig. 7 suplementar ).
Uma tabela SNP criada com MultiVCFAnalyzer v.0.85, contendo todas as posições variantes no conjunto de dados presente, foi filtrada para identificar diferenças SNP entre os genomas BSK001 e BSK003. As diferenças identificadas ( n = 20) foram então avaliadas com a ferramenta Java SNP_Evaluation 30 (data de compilação 13 de agosto de 2018; https://github.com/andreasKroepelin/SNP_Evaluation ). A tabela de variantes e os arquivos VCF de cada genoma foram usados como entrada para SNP_Evaluation. Além disso, cada variante privada identificada foi avaliada dentro de uma janela de 50 pb e foi considerada 'verdadeira' ao preencher os seguintes critérios estabelecidos em estudos publicados anteriormente 17 , 21 , 30 , 76: (1) nenhum sítio multi-alélico foi permitido dentro da janela avaliada, a menos que fossem consistentes com desaminação de aDNA (significado como substituições espúrias de C-para-T ou G-para-A); (2) a própria posição do SNP avaliada não era consistente com o dano de aDNA (nenhuma base sobreposta ao SNP foi reduzida por mapDamage v.2.0 (ref. 64 )); (3) não foram identificadas lacunas na cobertura genômica na janela avaliada; (4) leituras sobrepostas aos sítios SNP mostraram especificidade para o complexo Y. pseudotuberculosis quando rastreadas com BLASTn ( https://blast.ncbi.nlm.nih.gov/Blast.cgi ).
Finalmente, para obter resolução filogenética, os conjuntos de dados BSK001 e BSK003 Y. pestis foram concatenados. Realizamos concatenação de arquivos BAM, filtragem MALT 62 e reescalonamento de danos de aDNA (com mapDamage v.2.0 (ref. 64 )) conforme descrito na seção ' Processamento de dados pós-captura de Y. pestis '. Além disso, o conjunto de dados foi incluído na análise comparativa de SNP usando MultiVCFAnalyzer v.0.85 (ref. 74 ) conforme descrito acima. Finalmente, SNPs únicos foram avaliados com SNP_Evaluation 30 de acordo com os quatro critérios listados acima.
Reconstrução filogenética e estimativas de diversidade
A análise filogenética foi usada para explorar 233 genomas de Y. pestis como parte do moderno conjunto de dados comparativos. Um alinhamento de SNP produzido por MultiVCFAnalyzer v.0.85 (ref. 74 ) foi usado para construir uma árvore filogenética em MEGA7, usando a abordagem de máxima parcimônia com 95% de deleção parcial (6.032 SNPs). Dos 233 genomas modernos de Y. pestis no conjunto de dados atual, 30 exibiram extensos comprimentos de ramos privados (Fig. 12 suplementar). Tal efeito em filogenias bacterianas pode resultar tanto da verdadeira diversidade biológica quanto de artefatos técnicos associados à falsa incorporação de SNP durante a reconstrução computacional do genoma. Embora não possamos excluir a presença de várias cepas com taxas de mutação extremamente mais altas no conjunto de dados atual, estudos anteriores mostraram que cepas modernas de Y. pestis com perfis 'mutadores' são incomuns 16 , 36. Neste estudo, 27 dos 30 genomas que mostraram disparidades em suas contagens de SNP privados em comparação com o restante do conjunto de dados foram derivados de montagens para as quais a qualidade das chamadas de SNP não pôde ser avaliada (dados brutos indisponíveis). Como possíveis erros de montagem ou chamadas de SNP falso-positivos podem afetar inferências evolutivas e estimativas de diversidade, esses genomas foram excluídos de análises adicionais. Portanto, realizamos análise filogenética usando um subconjunto de 203 genomas modernos de Y. pestis (Tabela Complementar 13 ). A lista de genomas excluídos é a seguinte: 2.MED1_139 (ref. 19 ), 2.MED1_A-1809 (ref. 18 ), 2.MED1_A-1825 (ref. 19 ), 2.MED1_A-1920 (ref. 19 ) , 2.MED0_C-627 (ref.19 ), 2.MED1_M-1484 (ref. 19 ), 2.MED1_M-519 (ref. 19 ), 0.ANT5_A-1691 (ref. 18 ), 0.ANT5_A-1836 (ref. 18 ), 0.PE2_C -678 (ref. 77 ), 0.PE2_C-370 (ref. 77 ), 0.PE2_C-700 (ref. 77 ), 0.PE2_C-746 (ref. 77 ), 0.PE2_C-535 (ref. 77 ) ), 0.PE2_C-824 (ref. 77 ), 0.PE2_C-712 (ref. 77 ), 0.PE2b_G8786 (ref. 16 ), 0.PE4_I-3446 (ref. 78 ), 0.PE4_I-3517 ( ref. 78 ), 0.PE4t_A-1815 (ref. 18 ), 0.PE4_I-3447 (ref. 78 ), 0.PE4_I-3518 (ref.78 ), 0.PE4_I-3443 (ref. 78 ), 0.PE4_I-3442 (ref. 78 ), 0.PE4_I-3519 (ref. 78 ), 0.PE4_I-3516 (ref. 78 ), 0.PE4_I -3515 (ref. 78 ), 0.PE4_Microtus91001 (ref. 79 ), 0.PE5_I-2238 (ref. 80 ) e 0.PE7b_620024 (ref. 16 ).
Um alinhamento de SNP em todo o genoma consistindo em 203 genomas modernos e 48 históricos de Y. pestis (Tabela Complementar 14 ), bem como o genoma de Y. pseudotuberculosis IP32953, foi usado como entrada para construir uma filogenia de máxima probabilidade, incluindo 2.960 SNPs e até para 4% de dados perdidos. Realizamos análise filogenética com RAxML 81 v.8.2.9 usando o modelo de substituição reversível no tempo generalizado (GTR) com 4 categorias de taxa gama. Finalmente, 1.000 réplicas de bootstrap foram usadas para estimar o suporte do nó para a topologia de árvore resultante. Após a conclusão da execução, as filogenias de máxima verossimilhança foram visualizadas com FigTree v.1.4.4 ( http://tree.bio.ed.ac.uk/software/figtree/ ) e GrapeTree (v1.5.0) 50.
Para estimar a proporção da diversidade moderna de Y. pestis descendente de BSK001/003, usamos o pacote R picante v1.8.2 82 para calcular o MPD e FPD 83 da árvore de substituição de máxima verossimilhança reconstruída. As medidas feitas em um subconjunto da árvore correspondente ao subclado descendente de BSK001/003 (ramos 1–4) foram comparadas com a filogenia completa de Y. pestis . Em ambos os casos, apenas cepas modernas foram incluídas no cálculo. Usamos uma abordagem bootstrapping para avaliar a sensibilidade de nossos resultados em relação à amostragem e incerteza filogenética 84. Para cada uma das 1.000 árvores bootstrap RAxML, reamostramos aleatoriamente as cepas modernas com reposição e apenas mantivemos os galhos da árvore correspondentes às cepas amostradas. Medidas de diversidade foram realizadas para cada uma das árvores bootstrap reamostradas obtidas, a partir das quais foram derivadas estimativas medianas e intervalos de percentis de 95%.
Para avaliar o impacto potencial da amostragem desigual entre os ramos (os ramos 1-4 continham 130 cepas modernas, enquanto o ramo 0 continha apenas 73), repetimos a mesma análise, mas acrescentando uma etapa inicial destinada a equalizar o número de genomas em ambas as partes da árvore . Subamostramos os ramos 1–4 para o mesmo número de cepas que no ramo 0 usando agrupamento de sequências nos ramos 1–4 para obter subamostras representativas. Realizamos agrupamento hierárquico com base em distâncias filogenéticas pareadas (derivadas da árvore filogenética de máxima verossimilhança) e a árvore resultante foi cortada para definir 73 agrupamentos (funções hclust 85e cutree em R v.4.0.3). Para cada árvore bootstrap, os agrupamentos foram aleatoriamente reduzidos para uma linhagem, resultando em um número igual de linhagens entre o ramo 1-4 e o ramo 0. A reamostragem com substituição foi então aplicada como anteriormente para cada uma das árvores reduzidas antes de calcular as medidas de diversidade.
Análise de plasmídeo SNP
Para investigar uma possível variação genética entre os plasmídeos de genomas históricos, realizamos o mapeamento de leitura de BSK001, BSK003 e BSK001/003 com BWA, bem como a chamada de SNP com GATK v.3.5, conforme descrito na seção acima 'chamada de SNP, estimativas de heterozigosidade e SNP filtrar' contra cada um dos três plasmídeos de Y. pestis (pMT1: NC_003134.1; pPCP1: NC_003132.1; e pCD1: NC_003131.1). Em seguida, realizamos a chamada SNP comparativa usando o MultiVCFAnalyzer v0.85 (ref. 74 ) contra um conjunto de 46 Y. pestis históricosgenomas, bem como as cepas de referência modernas CO92, KIM5 e 0.PE4-Microtus91001. As variantes foram filtradas em genomas individuais usando SNP_Evaluation de acordo com critérios previamente definidos (consulte a seção 'Chamada de SNP, estimativas de heterozigosidade e filtragem de SNP'). No presente conjunto de dados, identificamos dez variantes em pCD1, oito em pMT1 e duas em pPCP1 (Tabela Complementar 15 ).
Análise filogenética calibrada no tempo
Para estimar o tempo para a divergência dos ramos 1-4 de Y. pestis usando os genomas BSK001/003 como um novo ponto de calibração, usamos um conjunto de dados compreendendo todos os genomas modernos dos ramos 1-4 usados para análise filogenética ( n = 130), genomas da linhagem ramificada ancestral 0.ANT3 ( n = 8) e todos os 29 genomas históricos (séculos XIV-XVIII) em nosso conjunto de dados representando genótipos únicos. Em casos de genomas idênticos, o genoma de maior cobertura foi escolhido para esta análise. Aplicamos um teste de relógio molecular usando um método de máxima verossimilhança no MEGA7 (ref. 86), usando um modelo de substituição GTR no qual as diferenças nas taxas evolutivas entre os sítios foram estimadas usando uma distribuição gama discreta com quatro categorias de taxa. Com base nesse teste do relógio molecular, a hipótese nula de taxas evolutivas iguais entre os ramos filogenéticos testados foi rejeitada, o que é consistente com estudos anteriores mostrando variação da taxa de substituição entre linhagens de Y. pestis 16 , 17 . Portanto, um modelo de relógio relaxado log-normal foi usado para todas as análises de datação molecular subsequentes.
Para a análise de datação molecular, usamos o framework estatístico Bayesiano BEAST2 v.6.6 (ref. 87 ). As idades de todos os isolados antigos foram usadas como pontos de calibração para construir uma filogenia calibrada no tempo com suas faixas etárias de radiocarbono ou contexto arqueológico definidas como anteriores uniformes (consulte a Tabela Suplementar 19 para todas as faixas etárias usadas). As idades de todos os isolados modernos foram fixadas em 0 anos antes do presente. Testamos uma série de árvores coalescentes prioritárias, como o tamanho constante coalescente, horizonte Bayesiano 88 e modelos de população exponencial, todos os quais foram usados ou testados em estudos genômicos de patógenos antigos anteriores 17 , 89 , 90 , 91. Também testamos a árvore do horizonte de nascimento-morte anterior, que ganhou força nos últimos anos 91 , 92 , 93 porque pode explicar variáveis epidemiológicas e modelar disparidades de amostragem ao longo do tempo 94 . Além disso, usamos jModelTest v.2.1.10 (ref. 95 ) para identificar o modelo de substituição de melhor ajuste para nosso conjunto de dados. O modelo de transversão indicado foi implementado em BEAUti usando um modelo GTR (4 categorias de taxa gama) e o parâmetro de taxa de substituição AG fixado em 1,0 (como indicado anteriormente 93 ). Todas as árvores prioritárias foram usadas em combinação com uma taxa de clock relaxada log-normal com uma distribuição a priori uniforme variando entre 1 × 10 −3 e 1 × 10−6 substituições por sítio por ano para o alinhamento SNP (1.405 sítios após uma deleção parcial de 95%), correspondendo a um intervalo de 3 × 10 −7 a 3 × 10 −10 em todo o genoma, que está dentro do intervalo de estimativas 17 . Como parte da configuração da topologia filogenética, todos os genomas dos ramos 1-4 (antigos e modernos), bem como a linhagem 0.ANT3, foram restringidos a serem clados monofiléticos independentes. Para o tamanho da população constante e os priors da árvore de população exponencial, todos os outros parâmetros foram definidos como padrão. Para a árvore do horizonte coalescente anterior, uma distribuição anterior de Jeffreys (1/ x) foi usado para os tamanhos de população e uma dimensão de 5 foi usada para permitir variações nos tamanhos de grupo e população ao longo do tempo, com um limite superior de 380.000 para o tamanho efetivo da população (padrão). Além disso, para a árvore do horizonte de nascimento-morte anterior, usamos uma prévia uniforme para a taxa de se tornar não infecciosa que variou entre 0,03 e 70, para considerar possíveis períodos infecciosos variando de 30 anos (infecções ao longo da vida em reservatórios de roedores 96 , 97 ) a 5 dias (período infeccioso médio para peste bubônica 98 ). Usamos uma distribuição beta anterior com média = 0,1 (alfa = 10,0, beta = 90,0) para a probabilidade de amostragem ρ no tempo 0 e uma distribuição uniforme variando entre 0 e 0,1 para a proporção de amostragem s. Para este último, foram permitidos dois turnos ao longo do tempo. Finalmente, permitiu-se que o número reprodutivo R variasse entre 0 e 4,0 usando uma longa distribuição normal anterior de mediana = 1,0 e dp = 0,7, que está dentro da faixa de estimativas anteriores para peste bubônica e pneumônica durante epidemias medievais 98 .
A adequação de todas as árvores prioritárias foi avaliada usando amostragem de caminho conforme implementado no pacote de seleção de modelo do BEAST2 v.6.6. A amostragem de caminho foi executada em 50 etapas, com 20 milhões de estados como o comprimento da cadeia para cada etapa. As probabilidades log-marginais resultantes favoreceram com 'forte suporte' 99 o modelo coalescente skyline para a presente análise (fator log Bayes = 8,35 quando comparado com o segundo melhor modelo) (Tabela Suplementar 20 ). Portanto, o modelo de skyline coalescente foi escolhido para análise posterior. Para avaliar o sinal temporal no presente conjunto de dados, usamos TempEst v.1.5.3 para estimar a distância raiz-ponta em relação às idades dos espécimes em uma análise de regressão linear 100 . Para TempEst, usamos uma árvore de máxima parcimônia calculada em MEGA7 (ref.86 ) no formato NEXUS. Além disso, usamos o ponto médio dos intervalos de datas arqueológicas ou de radiocarbono para todos os genomas antigos como datas de ponta. Todas as idades do genoma moderno foram definidas para 0 anos antes do presente. Os valores resultantes do coeficiente de correlação r (0,39) e R 2 (0,16) apoiaram a existência de um sinal temporal no presente conjunto de dados. Além disso, usamos a abordagem BETS 101para uma avaliação de sinal temporal que leva em consideração todos os parâmetros de análise. O BETS compara as estimativas de verossimilhança marginal (log) produzidas a partir de um modelo isócrono (todas as datas de amostragem definidas como 0 anos antes do presente) com um modelo heterócrono (incluindo tempos de amostragem reais). Como anteriormente, a amostragem de caminho foi executada em 50 etapas com 20 milhões de estados como o comprimento da cadeia para cada etapa. O fator (log)-Bayes estimado de 129,33 deu forte suporte ao modelo heterócrono; portanto, indicou a presença de um sinal temporal no presente conjunto de dados.
Para a análise de datação molecular usando uma configuração de modelo de horizonte coalescente, realizamos amostragem Monte Carlo de cadeia de Markov usando 2 cadeias independentes de 300 a 400 milhões de estados cada. Após a conclusão, as execuções foram combinadas usando LogCombiner v.2.6.7 e a convergência foi avaliada usando Tracer v.1.6 ( http://tree.bio.ed.ac.uk/software/tracer/ ) garantindo que os tamanhos de amostra efetivos fossem maiores de 200 para cada distribuição posterior estimada após um burn-in de 10%. Árvores de credibilidade máxima de clade foram construídas usando TreeAnnotator no pacote BEAST2 v.6.6 87com um burn-in de 10% e foram então visualizados no FigTree v.1.4.4. Em paralelo com a análise de datação molecular, realizamos uma amostragem da análise anterior para testar um possível overfitting da anterior aos dados. Realizamos amostragem Monte Carlo de cadeia de Markov para 2 cadeias independentes de 600 milhões de estados cada. Após a conclusão da corrida, as corridas foram combinadas e a convergência foi avaliada após um burn-in de 30%. Os resultados indicam que as distribuições posteriores do relógio relaxado log-normal não correlacionado e o tempo para as estimativas de ancestral comum mais recente não são concordantes com aqueles obtidos ao usar uma análise informada por dados (Fig. 13 suplementar ).
Como a maioria das estruturas filogenéticas Bayesianas (como BEAST2) são baseadas em árvores bifurcadas e, portanto, são pobres em resolver nós multifurcando, complementamos nossa abordagem usando TreeTime v.0.8.4 (ref. 35 ) para inferir uma filogenia calibrada no tempo usando um abordagem de máxima verossimilhança. O TreeTime demonstrou resolver politomias de maneira consistente com as datas de ponta da amostra. Geramos uma filogenia de máxima verossimilhança enraizada usando RAxML (Fig. 10 suplementar ) a partir do mesmo alinhamento de SNP usado para BEAST2 (95% de deleção parcial). A árvore de máxima verossimilhança foi então usada como entrada para TreeTime, que foi executado usando todas as datas de amostragem conhecidas para genomas modernos e o ponto médio da faixa etária para os genomas antigos (Tabela Suplementar 22). TreeTime foi executado usando a árvore coalescente Kingman antes com a configuração do horizonte. Um modelo de substituição apropriado foi escolhido para os dados usando a opção de inferência -gtr. A filogenia em escala de tempo foi inferida usando um relógio relaxado não correlacionado e com as opções de otimização de comprimento de ramificação, manter raiz e manter politomia. Além disso, os intervalos de tempo de divergência foram estimados a partir da árvore de maior verossimilhança usando a opção -confiança. As análises foram executadas usando um número máximo de 500 e 1.000 iterações (opção de número máximo de iterações) e produziram resultados consistentes. A árvore de tempo resultante pode ser encontrada na Fig. 11 Complementar .
Resumo do relatório
Mais informações sobre o projeto de pesquisa estão disponíveis no Nature Research Reporting Summary vinculado a este artigo.
Disponibilidade de dados
Os dados de sequência bruta produzidos neste estudo, as leituras alinhadas de Y. pestis após filtragem metagenômica e as leituras alinhadas por humanos estão disponíveis através do Arquivo Europeu de Nucleotídeos sob o número de acesso. PRJEB46734 . Mais dados estão disponíveis nas Informações Suplementares .
Disponibilidade do código
Nenhum código personalizado especializado foi usado para este estudo. Todos os softwares utilizados para as análises de dados neste estudo estão disponíveis publicamente.
Referências
Benedictow, OJ A Peste Negra, 1346-1353: a História Completa (Boydell & Brewer, 2004).
Clark, G. A Farewell to Alms: Uma Breve História Econômica do Mundo Vol. 25 (Princeton Univ. Press, 2008).
Pollitzer, R. Plague (Organização Mundial da Saúde, 1954).
Norris, J. Leste ou Oeste? A origem geográfica da Peste Negra. Touro. Hist. Med. 51 , 1-24 (1977).
PubMedGoogle ScholarDols, MW A Peste Negra no Oriente Médio (Princeton Univ. Press, 1979).
McNeill, WH Pragas e Povos (Âncora, 1976).
Campbell, BM A Grande Transição. Clima, doença e sociedade no mundo medieval tardio (Cambridge Univ. Press, 2016).
Stewart, J. Nestorian Missionary Enterprise: A História de uma Igreja em Chamas (T. & T. Clark, 1928).
Slavin, P. Morte à beira do lago: crise de mortalidade no início do século XIV na Ásia Central. J. Interdisciplinar. Hist. 50 , 59-90 (2019).
Google ScholarBos, KI et ai. Um esboço do genoma de Yersinia pestis de vítimas da Peste Negra. Natureza 478 , 506-510 (2011).
CASPubMedPubMed CentralArtigoGoogle ScholarVarlik, N. Plague and Empire in the Early Modern Mediterranean World (Cambridge Univ. Press, 2015).
Benedictow, OJ The Complete History of the Black Death (Boydell & Brewer, 2021).
Alfani, G. & Murphy, TE Pragas e epidemias letais no mundo pré-industrial. J. Eco. Hist. 77 , 314-343 (2017).
Google ScholarHerlihy, D. A Peste Negra e a Transformação do Oeste (Harvard Univ. Press, 1997).
Pamuk, Ş. A Peste Negra e as origens da 'Grande Divergência' na Europa, 1300-1600. EUR. Rev. Eco. Hist. 11 , 289-317 (2007).
Google ScholarCui, Y. et ai. Variações históricas na taxa de mutação em um patógeno epidêmico, Yersinia pestis . Proc. Natl Acad. Sci. EUA 110 , 577-582 (2013).
CASPubMedArtigoGoogle ScholarSpyrou, MA et ai. Filogeografia da segunda pandemia de peste revelada através da análise de genomas históricos de Yersinia pestis . Nat. Comum. 10 , 4470 (2019).
PubMedPubMed CentralArtigoCASGoogle ScholarEroshenko, GA et ai. As cepas de Yersinia pestis do antigo ramo filogenético 0.ANT estão amplamente espalhadas nos focos de peste de alta montanha do Quirguistão. PLoS ONE 12 , e0187230 (2017).
PubMed CentralArtigoCASGoogle ScholarKutyrev, VV et ai. Filogenia e classificação de Yersinia pestis através da lente de cepas dos focos de peste da Commonwealth of Independent States. Frente. Microbiol. 9 , 1106 (2018).
PubMed CentralArtigoGoogle ScholarGuelil, M. et ai. Uma síntese genômica e histórica da peste na Eurásia do século XVIII. Proc. Natl Acad. Sci. EUA 117 , 28328–28335 (2020).
PubMedPubMed CentralArtigoGoogle ScholarNamouchi, A. et ai. Abordagem integrativa usando genomas de Yersinia pestis para revisitar a paisagem histórica da peste durante o período medieval. Proc. Natl Acad. Sci. EUA 115 , E11790–E11797 (2018).
PubMedPubMed CentralArtigoGoogle ScholarHymes, R. Epílogo: uma hipótese sobre os primórdios da Ásia Oriental da politomia Yersinia pestis . Mediev. Globo 1 , 285-308 (2015).
Google ScholarChouin, G. Reflexões sobre a peste na história africana (14-19 c.). Afriques https://doi.org/10.4000/afriques.2228 (2018).
Fazlinejad, A. & Ahmadi, F. A Peste Negra no Irã, de acordo com relatos históricos iranianos dos séculos XIV a XV. J. Persianate Stud. 11 , 56-71 (2018).
Google ScholarBorsch, S. & Sabraa, T. Refugiados da Peste Negra: quantificando a migração rural para a peste e outros desastres ambientais. Ana Demogr. Hist. 134 , 63-93 (2017).
Green, MH As quatro pestes negras. Sou. Hist. Rev. 125 , 1601-1631 (2020).
Google ScholarChwolson, D. Syrisch-nestorianische Grabinschriften aus Semirjetschie, neue Folge (Commissionnaires de l'Académie Impériale des Sciences, 1897).
Fu, Q. et ai. Um humano moderno primitivo da Romênia com um ancestral neandertal recente. Natureza 524 , 216-219 (2015).
CASPubMedPubMed CentralArtigoGoogle ScholarHübler, R. et ai. HOPS: detecção e autenticação automatizadas de DNA de patógenos em vestígios arqueológicos. Genoma Biol. 20 , 280 (2019).
PubMed CentralArtigoCASGoogle ScholarKeller, M. et ai. Genomas antigos de Yersinia pestis de toda a Europa Ocidental revelam diversificação precoce durante a Primeira Pandemia (541-750). Proc. Natl Acad. Sci. EUA 116 , 12363–12372 (2019).
PubMedPubMed CentralArtigoGoogle ScholarWarinner, C. et ai. Uma estrutura robusta para arqueologia microbiana. Anu. Rev. Genom. Zumbir. Genet. 18 , 321-356 (2017).
ArtigoGoogle ScholarBos, KI et ai. Genomas de Yersinia pestis do século XVIII revelam a persistência a longo prazo de um foco histórico de peste. eLife 5 , e12994 (2016).
PubMed CentralArtigoGoogle ScholarGreen, MH Como um micróbio se torna uma pandemia: uma nova história da Peste Negra. Lancet Microbe 1 , e311–e312 (2020).
PubMedArtigoGoogle ScholarSpyrou, MA et ai. A análise de genomas de Yersinia pestis de 3800 anos sugere origem da Idade do Bronze para a peste bubônica. Nat. Comum. 9 , 2234 (2018).
PubMedPubMed CentralArtigoCASGoogle ScholarSagulenko, P., Puller, V. & Neher, RA TreeTime: análise filodinâmica de máxima probabilidade. Evolução do vírus. 4 , vex042 (2018).
PubMed CentralArtigoGoogle ScholarMorelli, G. et ai. O sequenciamento do genoma de Yersinia pestis identifica padrões de diversidade filogenética global. Nat. Genet. 42 , 1140-1143 (2010).
PubMedPubMed CentralArtigoGoogle ScholarZhou, Z. et ai. O guia do usuário EnteroBase, com estudos de caso sobre transmissões de Salmonella , filogenia de Yersinia pestis e diversidade genômica central de Escherichia . Genoma Res. 30 , 138-152 (2020).
PubMedPubMed CentralArtigoGoogle ScholarSusat, J. et ai. Um caçador-coletor de 5.000 anos já atormentado por Yersinia pestis . Cell Rep. 35 , 109278 (2021).
PubMedArtigoGoogle Scholarde Barros Damgaard, P. et ai. 137 genomas humanos antigos de todas as estepes da Eurásia. Natureza 557 , 369-374 (2018).
ArtigoCASGoogle ScholarAnisimov, AP, Lindler, LE & Pier, GB Diversidade intraespecífica de Yersinia pestis . Clin. Microbiol. Rev. 17 , 434–464 (2004).
PubMedPubMed CentralArtigoGoogle ScholarSpyrou, MA, Bos, KI, Herbig, A. & Krause, J. A genômica de patógenos antigos como uma ferramenta emergente para a pesquisa de doenças infecciosas. Nat. Rev. Genet. 20 , 323-340 (2019).
PubMedPubMed CentralArtigoGoogle ScholarFancy, NAG & Green, MH Plague e a queda de Bagdá (1258). Med. Hist. 65 , 157-177 (2021).
ArtigoGoogle ScholarMcCormick, M. Ratos, comunicações e peste: em direção a uma história ecológica. J. Interdisciplinar. Hist. 34 , 1-25 (2003).
Google ScholarPourhossein, B., Esmaeili, S., Gyuranecz, M. & Mostafavi, E. Tularemia e pesquisa de peste em roedores em uma zona de terremoto no sudeste do Irã. Epidemiol. Saúde 37 , e2015050 (2015).
PubMed CentralArtigoGoogle ScholarDennis, DT et ai. Manual da Peste: Epidemiologia, Distribuição, Vigilância e Controle (Organização Mundial da Saúde, 1999).
Schmid, BV et ai. Introdução da peste negra e sucessivas reintroduções da peste na Europa devido ao clima. Proc. Natl Acad. Sci. EUA 112 , 3020–3025 (2015).
CASPubMedPubMed CentralArtigoGoogle ScholarBarker, H. Colocando os cadáveres para descansar: grãos, embargos e Yersinia pestis no Mar Negro, 1346-48. Espéculo 96 , 97-126 (2021).
Google ScholarGómez, JM & Verdú, M. A teoria da rede pode explicar a vulnerabilidade dos assentamentos humanos medievais à pandemia da Peste Negra. Sci. Rep. 7 , 43467 (2017).
PubMedPubMed CentralArtigoGoogle ScholarLiu, X. A Rota da Seda na História Mundial (Oxford Univ. Press, 2010).
Zhou, Z. et ai. GrapeTree: visualização das principais relações genômicas entre 100.000 patógenos bacterianos. Genoma Res. 28 , 1395-1404 (2018).
PubMedPubMed CentralArtigoGoogle ScholarSistema de Informação Geográfica QGIS http://www.qgis.org (QGIS, 2022).
Neumann, GU, Andrades Valtuena, A., Fellows Yates, JA, Stahl, R. & Brandt, G. Amostragem de dentes da câmara pulpar interna para extração de DNA antigo V.2. protocol.io https://doi.org/10.17504/protocols.io.bqebmtan (2020).
Dabney, J. et ai. Sequência completa do genoma mitocondrial de um urso das cavernas do Pleistoceno Médio reconstruído a partir de fragmentos de DNA ultracurtos. Proc. Natl Acad. Sci. EUA 110 , 15758–15763 (2013).
CASPubMedPubMed CentralArtigoGoogle ScholarVelsko, I., Skourtanioti, E. & Brandt, G. Extração de DNA antigo de material esquelético. protocol.io https://doi.org/10.17504/protocols.io.baksicwe (2020).
Meyer, M. & Kircher, M. Preparação da biblioteca de sequenciamento Illumina para captura e sequenciamento de alvos altamente multiplexados. Porto da Primavera Fria. Protocolo 2010 , pdb.prot5448 (2010).
ArtigoGoogle ScholarRohland, N., Harney, E., Mallick, S., Nordenfelt, S. & Reich, D. Tratamento parcial com uracil-DNA-glicosilase para triagem de DNA antigo. Philos. Trans. R. Soc. Londres. B 370 , 20130624 (2015).
CASGoogle ScholarAron, F., Neumann, GU & Brandt, G. Preparação de biblioteca de DNA antigo de fita dupla tratada com metade de UDG para sequenciamento de ilumina. protocol.io https://doi.org/10.17504/protocols.io.bmh6k39e (2020).
Kircher, M., Sawyer, S. & Meyer, M. A indexação dupla supera as imprecisões no sequenciamento multiplex na plataforma Illumina. Res. de Ácidos Nucleicos. 40 , e3 (2012).
PubMedArtigoGoogle ScholarStahl, R. et ai. Indexação dupla de DNA de fita dupla da Illumina para DNA antigo. protocol.io https://doi.org/10.17504/protocols.io.bakticwn (2020).
Schubert, M., Lindgreen, S. & Orlando, L. AdapterRemoval v2: ajuste rápido do adaptador, identificação e mesclagem de leitura. BMC Res. Notas 9 , 88 (2016).
PubMed CentralArtigoGoogle ScholarO'Leary, NA et ai. Banco de dados de sequência de referência (RefSeq) no NCBI: status atual, expansão taxonômica e anotação funcional. Res. de Ácidos Nucleicos. 44 , D733–D745 (2016).
ArtigoCASGoogle ScholarVagene, Å. J. et ai. Genomas de Salmonella enterica de vítimas de uma grande epidemia do século XVI no México. Nat. Eco Evoluir 2 , 520-528 (2018).
ArtigoGoogle ScholarLi, H. et ai. O formato Sequence Alignment/Map e SAMtools. Bioinformática 25 , 2078–2079 (2009).
PubMed CentralArtigoCASGoogle ScholarJónsson, H., Ginolhac, A., Schubert, M., Johnson, P-LF & Orlando, L. mapDamage2.0: estimativas Bayesianas aproximadas rápidas de parâmetros de dano de DNA antigo. Bioinformática 29 , 1682-1684 (2013).
PubMed CentralArtigoCASGoogle ScholarGansauge, M.-T., Aximu-Petri, A., Nagel, S. & Meyer, M. Preparação manual e automatizada de bibliotecas de DNA de fita simples para o sequenciamento de DNA de restos biológicos antigos e outras fontes de DNA altamente degradado . Nat. Protocolo 15 , 2279-2300 (2020).
PubMedArtigoGoogle ScholarAndrades Valtueña, A. et al. A praga da Idade da Pedra e sua persistência na Eurásia. atual Biol. 27 , 3683-3691.e8 (2017).
ArtigoCASGoogle ScholarMathieson, I. et ai. Padrões de seleção de todo o genoma em 230 eurasianos antigos. Natureza 528 , 499-503 (2015).
CASPubMedPubMed CentralArtigoGoogle ScholarFu, Q. et ai. Análise de DNA de um humano moderno da caverna Tianyuan, China. Proc. Natl Acad. Sci. EUA 110 , 2223-2227 (2013).
CASPubMedPubMed CentralArtigoGoogle ScholarHaak, W. et ai. A migração maciça da estepe foi uma fonte de línguas indo-européias na Europa. Natureza 522 , 207-211 (2015).
CASPubMedPubMed CentralArtigoGoogle ScholarPeltzer, A. et ai. EAGER: reconstrução eficiente do genoma antigo. Genoma Biol. 17 , 60 (2016).
PubMed CentralArtigoCASGoogle ScholarSchuenemann, VJ et ai. Enriquecimento direcionado de patógenos antigos produzindo o plasmídeo pPCP1 de Yersinia pestis de vítimas da Peste Negra. Proc. Natl Acad. Sci. EUA 108 , E746–E752 (2011).
PubMedPubMed CentralArtigoGoogle ScholarHuson, DH et ai. MEGAN Community Edition - exploração interativa e análise de dados de sequenciamento de microbioma em larga escala. Computação PLoS. Biol. 12 , e1004957 (2016).
PubMed CentralArtigoCASGoogle ScholarDe Pristo, MA et al. Uma estrutura para descoberta de variação e genotipagem usando dados de sequenciamento de DNA de última geração. Nat. Genet. 43 , 491-498 (2011).
PubMedPubMed CentralArtigoGoogle ScholarBos, KI et ai. Genomas micobacterianos pré-colombianos revelam focas como fonte de tuberculose humana do Novo Mundo. Natureza 514 , 494-497 (2014).
CASPubMedPubMed CentralArtigoGoogle ScholarEquipe R. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2019).
Feldman, M. et ai. Um genoma de Yersinia pestis de alta cobertura de uma vítima da peste justiniana do século VI. Mol. Biol. Evoluir 33 , 2911-2923 (2016).
PubMedPubMed CentralArtigoGoogle ScholarKislichkina, AA et ai. Oito conjuntos de genoma completo de Yersinia pestis subsp. microtus bv. Caucasica isolada do foco de praga da ratazana comum ( Microtus arvalis ) no Daguestão, Rússia. Genoma Anunciado. 5 , e00847-17 (2017).
PubMed CentralArtigoGoogle ScholarKislichkina, AA et ai. Nove conjuntos de genoma completo de Yersinia pestis subsp. microtus bv. Cepas Altaica isoladas do foco de praga natural da Montanha Altai (nº 36) na Rússia. Genoma Anunciado. 6 , e01440-17 (2018).
PubMed CentralGoogle ScholarZhou, D. et ai. Genética das variações metabólicas entre biovars de Yersinia pestis e a proposta de um novo biovar, microtus . J. Bacteriol. 186 , 5147-5152 (2004).
PubMedPubMed CentralArtigoGoogle ScholarKislichkina, AA et ai. Seis conjuntos de genoma completo de Yersinia pestis subsp. microtus bv. Cepas de Ulegeica (filogrupo o.PE5) isoladas de focos de peste natural da Mongólia. Genoma Anunciado. 6 , e00536-18 (2018).
PubMed CentralGoogle ScholarStamatakis, A. RAxML versão 8: uma ferramenta para análise filogenética e pós-análise de grandes filogenias. Bioinformática 30 , 1312-1313 (2014).
PubMedPubMed CentralArtigoGoogle ScholarKembel, SW et ai. Picante: ferramentas R para integrar filogenias e ecologia. Bioinformática 26 , 1463-1464 (2010).
PubMedArtigoGoogle ScholarFé, DP Avaliação da conservação e diversidade filogenética. Biol. Conserv. 61 , 1-10 (1992).
Google ScholarEfron, B. & Tibshirani, R. Métodos de Bootstrap para erros padrão, intervalos de confiança e outras medidas de precisão estatística. Estado. Sci. 1 , 54-74 (1986).
MATEMÁTICAGoogle ScholarMüllner, D. Fastcluster: rotinas rápidas de agrupamento hierárquico e aglomerativo para R e Python. J. Stat. Softw. 53 , 1-18 (2013).
Google ScholarKumar, S., Stecher, G. & Tamura, K. MEGA7: Análise de Genética Evolutiva Molecular versão 7.0 para conjuntos de dados maiores. Mol. Biol. Evoluir 33 , 1870-1874 (2016).
PubMedPubMed CentralArtigoGoogle ScholarBouckaert, R. et ai. BEAST 2.5: uma plataforma de software avançada para análise evolutiva Bayesiana. Computação PLoS. Biol. 15 , e1006650 (2019).
PubMedPubMed CentralArtigoGoogle ScholarDrummond, AJ, Rambaut, A., Shapiro, B. & Pybus, OG Bayesian inferência coalescente de dinâmica populacional passada de sequências moleculares. Mol. Biol. Evoluir 22 , 1185-1192 (2005).
PubMedArtigoGoogle ScholarMühlemann, B. et ai. Diversas cepas de vírus da varíola (varíola) foram difundidas no norte da Europa na Era Viking. Science 369 , eaaw8977 (2020).
ArtigoCASGoogle ScholarGiffin, K. et ai. Um genoma treponêmico de uma vítima histórica da peste apóia um recente surgimento de bouba e sua presença na Europa do século XV. Sci. Rep. 10 , 9499 (2020).
CASPubMedPubMed CentralArtigoGoogle ScholarKocher, A. et ai. Dez milênios de evolução do vírus da hepatite B. Ciência 374 , 182-188 (2021).
CASPubMedArtigoGoogle ScholarChave, FM et al. A emergência de Salmonella enterica adaptada ao homem está ligada ao processo de neolitização. Nat. Eco Evoluir 4 , 324-333 (2020).
PubMed CentralArtigoGoogle ScholarSabin, S. et ai. Um genoma de Mycobacterium tuberculosis do século XVII suporta um surgimento neolítico do complexo Mycobacterium tuberculosis . Genoma Biol. 21 , 201 (2020).
PubMedPubMed CentralArtigoGoogle ScholarStadler, T., Kühnert, D., Bonhoeffer, S. & Drummond, AJ O gráfico do horizonte de nascimento-morte revela mudanças temporais na propagação da epidemia no HIV e no vírus da hepatite C (HCV). Proc. Natl Acad. Sci. EUA 110 , 228–233 (2013).
CASPubMedArtigoGoogle ScholarDarriba, D., Taboada, GL, Doallo, R. & Posada, D. jModelTest 2: mais modelos, novas heurísticas e computação paralela. Nat. Métodos 9 , 772 (2012).
PubMedPubMed CentralArtigoGoogle ScholarGorbunova, V., Bozzella, MJ & Seluanov, A. Roedores para estudos comparativos de envelhecimento: de camundongos a castores. Idade (Dordr.) 30 , 111–119 (2008).
Google ScholarMahmoudi, A. et ai. Espécies de reservatórios de peste em todo o mundo. Integr. Zool. 16 , 820-833 (2021).
ArtigoGoogle ScholarDean, KR et ai. Ectoparasitas humanos e a propagação da peste na Europa durante a Segunda Pandemia. Proc. Natl Acad. Sci. EUA 115 , 1304–1309 (2018).
PubMedPubMed CentralArtigoGoogle ScholarKass, RE & Raftery, AE Fatores de Bayes. Geléia. Estado. Associação 90 , 773-795 (1995).
MATEMÁTICAArtigoGoogle ScholarRambaut, A., Lam, TT, Max Carvalho, L. & Pybus, OG Explorando a estrutura temporal de sequências heterócronas usando TempEst (anteriormente Path-O-Gen). Evolução do vírus. 2 , ver007 (2016).
PubMed CentralArtigoGoogle ScholarDuchene, S. et ai. Avaliação Bayesiana do sinal temporal em populações em evolução mensurável. Mol. Biol. Evoluir 37 , 3363-3379 (2020).
PubMedArtigoGoogle Scholar
Reconhecimentos
Agradecemos a A. Herbig, G. Neumann e A. Andrades Valtueña e todos os outros membros dos grupos de trabalho de Paleopatologia Molecular e Patogenômica Computacional do Instituto Max Planck de Antropologia Evolutiva pelas discussões úteis ao longo deste estudo; C. Posth e R. Barquera pelos comentários sobre as primeiras versões do manuscrito; T. Hermes pelas discussões úteis durante a revisão deste estudo; M. O'Reilly pelo suporte gráfico; RI Tukhbatova, N. Martins, F. Aron, A. Wissgott, R. Radzeviciute e G. Brandt no Instituto Max Planck para a Ciência da História Humana em Jena, bem como S. Nagel no Instituto Max Planck de Antropologia Evolutiva em Leipzig pelo apoio laboratorial; K. Prüfer e S. Clayton pela assistência computacional. A datação por radiocarbono ocorreu no Curt-Engelhorn-Zentrum Archäometrie em Mannheim, Alemanha; agradecemos a R. Freidrich por ajudar na interpretação dos resultados da datação por radiocarbono. Além disso, agradecemos a VI Selezneva do Museu de Antropologia e Etnografia Pedro, o Grande, pela ajuda inicial com a amostragem de espécimes e N. Smelova, atualmente na Universidade de Oslo, por fornecer informações contextuais essenciais durante a fase inicial deste projeto. Este projeto recebeu financiamento do Conselho Europeu de Pesquisa no âmbito do programa de pesquisa e inovação Horizonte 2020 da União Europeia sob a concessão no. 771234 (PALEORIDER para WH). LM e LD foram apoiados pela subvenção no. AP08856654 do Ministério da Educação e Ciência da República do Cazaquistão. MAS, GAGR, AK, KIB, DK e JK também foram apoiados pela Max Planck Society. Freidrich por ajudar na interpretação dos resultados da datação por radiocarbono. Além disso, agradecemos a VI Selezneva do Museu de Antropologia e Etnografia Pedro, o Grande, pela ajuda inicial com a amostragem de espécimes e N. Smelova, atualmente na Universidade de Oslo, por fornecer informações contextuais essenciais durante a fase inicial deste projeto. Este projeto recebeu financiamento do Conselho Europeu de Pesquisa no âmbito do programa de pesquisa e inovação Horizonte 2020 da União Europeia sob a concessão no. 771234 (PALEORIDER para WH). LM e LD foram apoiados pela subvenção no. AP08856654 do Ministério da Educação e Ciência da República do Cazaquistão. MAS, GAGR, AK, KIB, DK e JK também foram apoiados pela Max Planck Society. Freidrich por ajudar na interpretação dos resultados da datação por radiocarbono. Além disso, agradecemos a VI Selezneva do Museu de Antropologia e Etnografia Pedro, o Grande, pela ajuda inicial com a amostragem de espécimes e N. Smelova, atualmente na Universidade de Oslo, por fornecer informações contextuais essenciais durante a fase inicial deste projeto. Este projeto recebeu financiamento do Conselho Europeu de Pesquisa no âmbito do programa de pesquisa e inovação Horizonte 2020 da União Europeia sob a concessão no. 771234 (PALEORIDER para WH). LM e LD foram apoiados pela subvenção no. AP08856654 do Ministério da Educação e Ciência da República do Cazaquistão. MAS, GAGR, AK, KIB, DK e JK também foram apoiados pela Max Planck Society. Selezneva do Museu de Antropologia e Etnografia Pedro, o Grande, pela ajuda inicial com a amostragem de espécimes e N. Smelova, atualmente na Universidade de Oslo, por fornecer informações contextuais essenciais durante a fase inicial deste projeto. Este projeto recebeu financiamento do Conselho Europeu de Pesquisa no âmbito do programa de pesquisa e inovação Horizonte 2020 da União Europeia sob a concessão no. 771234 (PALEORIDER para WH). LM e LD foram apoiados pela subvenção no. AP08856654 do Ministério da Educação e Ciência da República do Cazaquistão. MAS, GAGR, AK, KIB, DK e JK também foram apoiados pela Max Planck Society. Selezneva do Museu de Antropologia e Etnografia Pedro, o Grande, pela ajuda inicial com a amostragem de espécimes e N. Smelova, atualmente na Universidade de Oslo, por fornecer informações contextuais essenciais durante a fase inicial deste projeto. Este projeto recebeu financiamento do Conselho Europeu de Pesquisa no âmbito do programa de pesquisa e inovação Horizonte 2020 da União Europeia sob a concessão no. 771234 (PALEORIDER para WH). LM e LD foram apoiados pela subvenção no. AP08856654 do Ministério da Educação e Ciência da República do Cazaquistão. MAS, GAGR, AK, KIB, DK e JK também foram apoiados pela Max Planck Society. por fornecer informações contextuais essenciais durante a fase inicial deste projeto. Este projeto recebeu financiamento do Conselho Europeu de Pesquisa no âmbito do programa de pesquisa e inovação Horizonte 2020 da União Europeia sob a concessão no. 771234 (PALEORIDER para WH). LM e LD foram apoiados pela subvenção no. AP08856654 do Ministério da Educação e Ciência da República do Cazaquistão. MAS, GAGR, AK, KIB, DK e JK também foram apoiados pela Max Planck Society. por fornecer informações contextuais essenciais durante a fase inicial deste projeto. Este projeto recebeu financiamento do Conselho Europeu de Pesquisa no âmbito do programa de pesquisa e inovação Horizonte 2020 da União Europeia sob a concessão no. 771234 (PALEORIDER para WH). LM e LD foram apoiados pela subvenção no. AP08856654 do Ministério da Educação e Ciência da República do Cazaquistão. MAS, GAGR, AK, KIB, DK e JK também foram apoiados pela Max Planck Society.
Financiamento
Financiamento de acesso aberto fornecido pela Max Planck Society.
Informação sobre o autor
Autores e Afiliações
Contribuições
MAS, PS e JK conceberam e conduziram a investigação. MAS, KIB, PS e JK desenharam o estudo. MAS e LM realizaram o trabalho de laboratório. MAS e AK realizaram a análise dos dados genômicos bacterianos. MAS, AK e DK realizaram a análise de datação molecular. O GAGR realizou a análise genética da população humana. P.-GB e PS reuniram, analisaram e traduziram as informações de contexto histórico, arqueológico e epigráfico. AB e VIK forneceram acesso ao material arqueológico e informações contextuais. MAS, AK, LD, KIB, DK, WH, PS e JK auxiliaram na interpretação dos resultados. MAS e PS escreveram o artigo com contribuições de todos os coautores.
Autores correspondentes
Declarações de ética
Interesses competitivos
Os autores declaram não haver interesses conflitantes.
Revisão por pares
Informações de revisão por pares
A Nature agradece a Ludovic Orlando, Mark Achtman e os outros revisores anônimos por sua contribuição para a revisão por pares deste trabalho. Relatórios de revisores estão disponíveis.
Informação adicional
Nota do editor Springer Nature permanece neutro em relação a reivindicações jurisdicionais em mapas publicados e afiliações institucionais.
Figuras e tabelas de dados estendidas
Dados estendidos Fig. 1 Imagens de lápides disponíveis de Kara-Djigach.
a–c , Imagens de lápides disponíveis de indivíduos investigados como parte deste estudo. Para obter uma tradução das inscrições da lápide, consulte as descrições individuais em Informações Complementares 2 . As datas das lápides são as seguintes: Sepultura 9 (1338-9 CE), Sepultura 19/20 (1338-9 CE), Sepultura 6 (ano não inscrito). Créditos das imagens para P.-G. Borbone. d+e , lápides identificadas em Kara-Djigach contendo inscrições que indicam pestilências, datando dos anos 1338 e 1339 EC. Essas lápides não correspondem a indivíduos analisados em nosso conjunto de dados de aDNA. A lápide original no painel d (sem inscrição traçada) é mostrada na Fig. 1 . Traduções completas estão disponíveis em Informações Complementares 2.
Dados estendidos Fig. 2 Freqüências de substituição de danos no DNA antigo para todas as bibliotecas capturadas de Y. pestis .
As frequências de substituição C-para-T características da desaminação post-mortem de DNA antigo são mostradas para as extremidades 5' de leituras sequenciadas alinhadas contra o genoma de referência CO92 Y. pestis (NC_003143.1).
Dados estendidos Fig. 3 Avaliação de BSK007 após a captura de todo o genoma de Y. pestis .
a , Distribuição de cobertura pós-captura de BSK007 ao longo do cromossomo Y. pestis CO92. A cobertura média foi estimada em todo o genoma em janelas de 4.000 pb. A linha cinza pontilhada indica a cobertura média em todo o genoma (0,125 vezes). b , gráficos de Krona mostrando a classificação taxonômica de BSK007 lê mapeamento contra todos os Y. pestisElementos CO92 (cromossoma NC_003143.1, pMT1 NC_003134.1, pPCP1 NC_003132.1 e pCD1 NC_003131.1). Os números entre colchetes ao lado das designações dos elementos correspondem ao número de leituras atribuídas no MALT. As cores dos setores Krona representam diferentes níveis taxonômicos e sua completude é proporcional à abundância relativa de leituras resumidas em cada nó taxonômico correspondente. As porcentagens mostradas indicam a proporção em nível de espécie (círculo mais externo) de reads alinhados a taxa diferentes de Y. pestis e Y. pseudotuberculosis .
Dados estendidos Fig. 4 Comprimento de leitura e distribuição de danos no DNA antigo de regiões SNP contaminantes no conjunto de dados BSK001/003 combinado antes da filtragem metagenômica.
Distribuições de comprimento sobrepostas de mapeamento de leituras contra o CO92 Y. pestisgenoma de referência, calculado para todo o conjunto de dados (cinza), bem como para 117 regiões ao redor de SNPs supostamente contaminantes (azul). As regiões foram extraídas dentro de uma janela de 150 pb ao redor de cada SNP supostamente contaminante. As linhas pontilhadas representam os comprimentos médios dos fragmentos para todo o conjunto de dados e para as 117 regiões SNP supostamente contaminantes em cinza e azul, respectivamente. As leituras que compreendem regiões SNP contaminantes mostram uma distribuição de comprimento distinta em comparação com a observada em todo o genoma BSK001/003, com uma mudança acentuada para comprimentos de leitura mais longos. O pico de comprimento de fragmento de 76 bp representa o comprimento de leitura mais alto possível de leituras sequenciadas de extremidade única, que compreendiam a maioria dos dados dentro do presente conjunto de dados.
Dados estendidos Fig. 5 Comparações filogenéticas entre BSK001/003 contra a diversidade de Y. pestis antiga e moderna.
Árvore filogenética de máxima verossimilhança de comprimento total usando 1.000 iterações de bootstrap para estimar o suporte do nó e visualizada usando o FigTree v1.4.4. A árvore é construída com 203 genomas modernos e 48 históricos de Y. pestis , e é baseada em 2.960 SNPs (96% deleção parcial). A escala denota o número de substituições por sítio genômico.
Dados estendidos Fig. 6 Avaliação de SNPs de diagnóstico filogeneticamente em genomas de Y. pestis do século XIV.
As chamadas variantes estimadas foram recuperadas de uma tabela SNP compreendendo 203 genomas modernos e 48 históricos de Y. pestis (conjunto de dados completo contém 3.533 SNPs). As barras de erro indicam incerteza devido à presença de dados ausentes (Ns) nas chamadas variantes dos respectivos genomas.
Dados estendidos Fig. 7 Comparação de medidas de diversidade filogenética calculadas para a filogenia completa de Y. pestis moderna contra o subclado Branch 1-4.
Os painéis da esquerda mostram árvores de substituição de probabilidade máxima considerando apenas genomas de Y. pestis existentes , anotados com base nos clados comparados e seu índice de diversidade filogenética de Faith (FPD) correspondente. Os ramos filogenéticos considerados para o cálculo do Ramo 1-4 FPD são mostrados em verde (conjunto de dados completo) e vermelho (conjunto de dados subamostrado). Os painéis da direita mostram gráficos de violino indicando a distribuição e a média das distâncias filogenéticas aos pares com base em árvores de máxima verossimilhança (MPD). As estimativas medianas e os intervalos percentuais de 95% foram derivados das árvores de bootstrap reamostradas (1.000 iterações de bootstrap). Os pontos dentro dos gráficos de violino indicam a distância filogenética média estimada para todos os conjuntos de dados.
Dados Estendidos Fig. 8 Relações filogenéticas de linhagens 0.ANT.
Árvore filogenética de máxima probabilidade com base em 2.441 posições variantes do genoma (todos os SNPs). A árvore foi construída para indicar as relações genéticas entre todos os genomas 0.ANT existentes publicados anteriormente e BSK001/003. A escala denota o número de substituições por sítio genômico. O suporte a nós foi determinado por 1.000 iterações de bootstrap.
Informação suplementar
Informação suplementar
Seções Suplementares 1–4 e Referências.
Nenhum comentário:
Postar um comentário
Observação: somente um membro deste blog pode postar um comentário.