Novas
pesquisas genéticas mostram que o DNA e as evidências arqueológicas
estão alinhados com a "longa cronologia" do povoamento da Austrália.
Mapa de Sunda, Sahul e do Pacífico Ocidental,
com setas indicando possíveis rotas de migração para o norte e para o
sul, sugeridas por análises genéticas. (Crédito da imagem: Helen Farr e Erich Fisher)
Um novo estudo com quase
2.500 genomas pode finalmente ter resolvido o debate sobre quando os
humanos modernos chegaram à Austrália. Usando um banco de dados
diversificado de DNA
de povos aborígenes antigos e contemporâneos de toda a Oceania,
pesquisadores determinaram que os primeiros habitantes do norte da
Austrália começaram a se estabelecer há 60.000 anos e que chegaram por
duas rotas distintas.
Especialistas debatem há muito tempo a data da chegada dos primeiros humanos à Austrália , um feito que exigiu a invenção de embarcações. Enquanto alguns pesquisadores usaram modelos genéticos para sustentar uma "cronologia curta" de 47.000 a 51.000 anos atrás para a chegada, outros reuniram evidências arqueológicas
e conhecimento aborígine em apoio à "cronologia longa", na qual as
primeiras chegadas ocorreram entre 60.000 e 65.000 anos atrás.
No novo estudo, publicado na sexta-feira (28 de novembro) na revista Science Advances
, pesquisadores analisaram um conjunto de dados "sem precedentes" de
2.456 genomas humanos para responder à questão de quando os humanos
migraram de Sunda (a antiga massa de terra, também conhecida como
Sundalândia, que incluía o que hoje são a Indonésia, as Filipinas e a
Península Malaia) para Sahul (um paleocontinente que incluía a Austrália, a Tasmânia e a Nova Guiné modernas).
"Este é o estudo genético
mais abrangente até o momento sobre essa questão e oferece forte apoio à
cronologia longa em vez da cronologia curta", disse o coautor do
estudo, Martin Richards , arqueogeneticista da Universidade de Huddersfield, no Reino Unido, em um e-mail para a Live Science.
A
análise da equipe também revelou dois grupos distintos de pessoas que
chegaram por rotas do norte e do sul. "Essa conclusão se encaixa muito
bem com as evidências arqueológicas e oceanográficas/paleoclimáticas de
uma entrada em Sahul por volta de 60.000 anos atrás", disse Richards.
Para chegar às suas conclusões, os pesquisadores utilizaram uma abordagem de relógio molecular
, que pressupõe que as mutações nas sequências de DNA ocorrem a uma
taxa relativamente constante ao longo do tempo. Ao analisar as
diferenças entre duas sequências de DNA, os pesquisadores podem estimar
quando essas sequências divergiram uma da outra.
No estudo, a
equipe de pesquisa utilizou diversos métodos estatísticos para analisar o
DNA mitocondrial (transmitido pela linhagem materna) e os dados do
cromossomo Y (transmitido pela linhagem paterna). Todos os seus modelos
estatísticos convergiram para uma data de aproximadamente 60.000 anos
atrás para o povoamento do norte da Austrália.
Mas os dados genéticos também revelaram dois assentamentos distintos
aproximadamente na mesma época. Um grupo de pessoas chegou à Austrália
através do sul de Sunda (as ilhas indonésias), enquanto outro veio do
norte de Sunda (o arquipélago filipino).
Os dois grupos faziam
inicialmente parte da mesma população que saiu da África há cerca de
70.000 a 80.000 anos, disse Richards, e "acreditamos que eles se
separaram durante a dispersão para o leste, no sul ou sudeste da Ásia",
possivelmente de 10.000 a 20.000 anos antes de chegarem à Austrália.
"Nossos
resultados indicam que os aborígenes australianos, juntamente com os
habitantes da Nova Guiné, possuem a ancestralidade ininterrupta mais
antiga de qualquer grupo de pessoas fora da África", disse Richards.
Ao longo do caminho, esses primeiros pioneiros humanos provavelmente se miscigenaram com humanos arcaicos, como o Homo longi , o H. luzonensis e até mesmo o "hobbit" H. floresiensis
, de acordo com Richards, mas atualmente não está claro até que ponto
os humanos modernos interagiram com os povos arcaicos na região.
Adam Brumm
, um arqueólogo da Universidade Griffith, na Austrália, que não
participou do estudo, disse ao Live Science por e-mail que a pesquisa
apoia a ideia de que os primeiros movimentos humanos tiveram um papel
crucial no povoamento inicial de Sahul. "Eu apostaria, se tivesse
dinheiro, no modelo da 'cronologia longa'", disse Brumm.
Este
estudo genético tem amplas implicações para a antiguidade dos povos
aborígenes na Austrália. "Muitos aborígenes australianos e habitantes
das Ilhas do Estreito de Torres entendem que sempre estiveram nesta
terra", disse a coautora do estudo, Helen Farr , arqueóloga da Universidade de Southampton, no Reino Unido, em um e-mail para a Live Science.
"Esses
dados comprovam uma herança cultural muito rica para essas
comunidades", disse Farr, "e revelam os laços estreitos que as pessoas
mantêm com a Terra e o Mar há pelo menos 60.000 anos". Mas também
demonstram que o conhecimento e as habilidades de navegação, que não são
encontrados no registro arqueológico, foram essenciais para a
sobrevivência dos primeiros humanos.
Nota do editor: Este
artigo foi atualizado na segunda-feira (1º de dezembro) às 4h30 (horário
do leste dos EUA) para corrigir um erro na citação original de Helen
Farr, que dizia "Os aborígenes acreditam que sempre estiveram na Terra"
para "Os aborígenes australianos e os habitantes das Ilhas do Estreito
de Torres entendem que sempre estiveram na Terra".
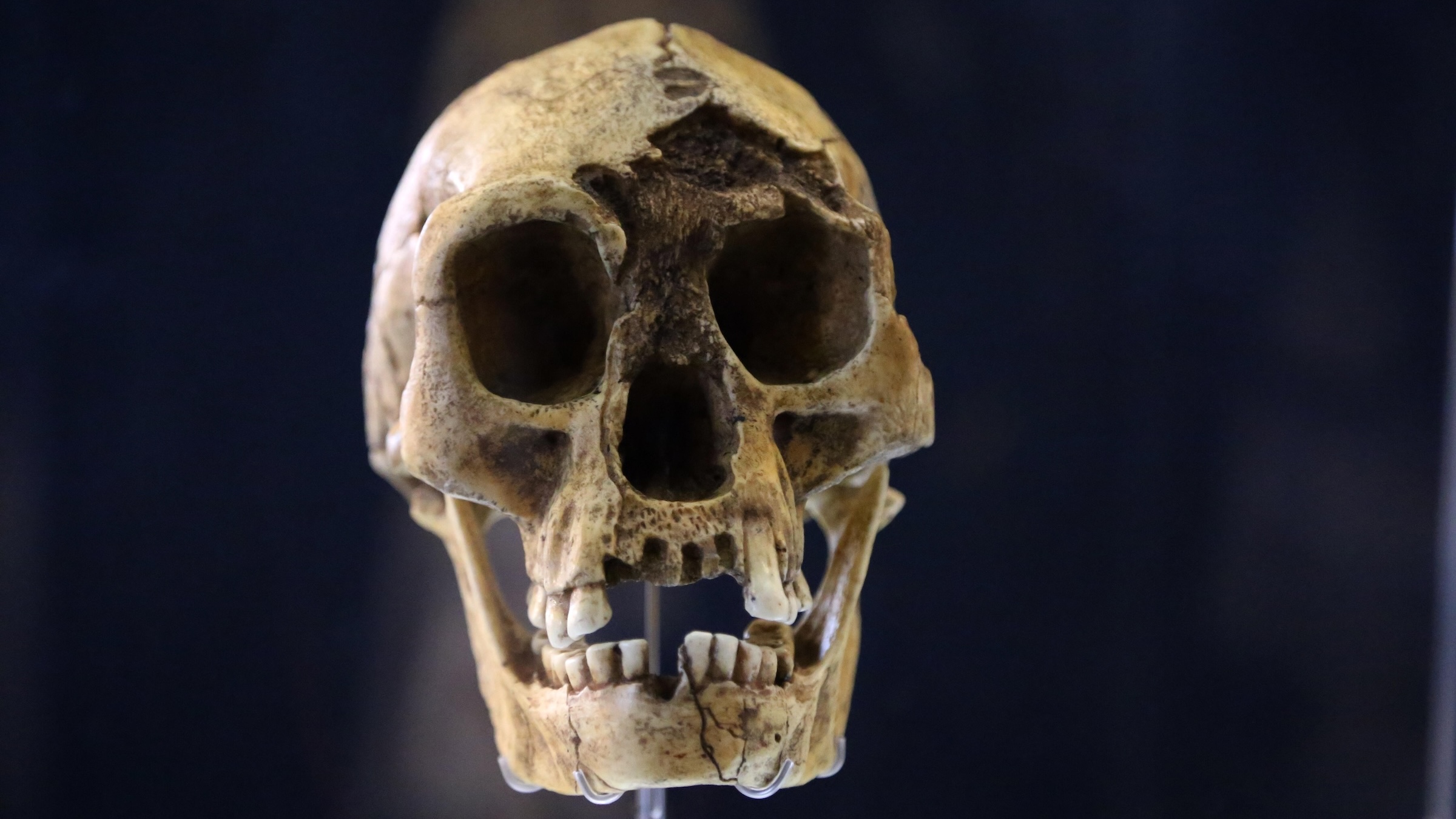
Uma nova pesquisa sugere que os 'hobbits' podem ter desaparecido quando a seca os obrigou a competir com os humanos modernos.
Uma
redução nas chuvas pode ter desempenhado um papel considerável na
extinção do Homo floresiensis, a espécie humana arcaica apelidada de
"hobbit", segundo um novo estudo.
Crânio de Homo floresiensis , também conhecido como "hobbit". (Crédito da imagem: Lanmas via Alamy)
O Homo floresiensis — uma pequena espécie humana antiga apelidada de "hobbit"
— pode ter sido extinto há cerca de 50.000 anos devido à diminuição dos
níveis de chuva, que reduziu a disponibilidade de presas para a caça.
Isso pode tê-los forçado a migrar para áreas onde competiam com os
humanos modernos, sugere uma nova pesquisa.
A
escassez de chuvas não teria sido o único motivo para a extinção dessas
espécies, observou a equipe. Uma erupção vulcânica ocorrida há cerca de
50.000 anos também pode ter sido um fator significativo para o seu
desaparecimento.
Até
o momento, fósseis do hobbit foram encontrados em apenas uma caverna,
conhecida como Liang Bua, na ilha de Flores, na Indonésia. Desde que a
descoberta do *H. floresiensis* foi relatada publicamente pela primeira vez em 2004 , os cientistas têm tentado determinar como essa espécie diminuta vivia e por que foi extinta.
Agora, em um artigo publicado na segunda-feira (8 de dezembro) no periódico Communications Earth & Environment
, cientistas relatam que a precipitação na ilha parece ter diminuído
consideravelmente antes de 50.000 anos atrás. Eles também descobriram
que a população de Stegodon , um gênero de parente extinto do
elefante que os hobbits caçavam, também diminuiu antes de desaparecer
de Flores há cerca de 50.000 anos.
Para
determinar como a precipitação na ilha mudou, a equipe estudou uma
estalagmite de Liang Luar, uma caverna em Flores próxima a Liang Bua. As estalagmites
crescem quando a água evapora e forma carbonato de cálcio. O novo
crescimento também contém pequenas quantidades de outros minerais, como
magnésio. As estalagmites não crescem tão rápido durante períodos de
escassez de água, e o crescimento que ocorre tende a ter menos carbonato
de cálcio e mais magnésio, observaram os pesquisadores em seu artigo.
Isso significa que, ao medir a proporção de magnésio para carbonato de
cálcio, a equipe pode determinar quando a precipitação diminuiu ou
aumentou e em que medida.
Os pesquisadores descobriram que a
precipitação média anual diminuiu de 1.560 milímetros (61,4 polegadas)
há 76.000 anos para 990 milímetros (40 polegadas) há 61.000 anos. A ilha
continuou com esse nível reduzido de precipitação até 50.000 anos
atrás. Nesse ponto, houve uma erupção em um vulcão próximo, e uma camada
de rochas ejetadas cobriu a ilha.
Ao analisar os restos de dentes de estegodonte
, a equipe descobriu que o número desses animais diminuiu na ilha entre
61.000 e 50.000 anos atrás, antes de desaparecerem após a erupção. Os
pesquisadores acreditam que a redução das chuvas levou à diminuição das populações de estegodonte , tornando a vida mais difícil para os hobbits, já que esses animais constituíam uma parte importante de sua dieta.
Com a diminuição das chuvas, as populações de Stegodon podem ter migrado para o litoral da ilha, com os hobbits seguindo-as.
"Suspeitamos que, se a população de Stegodon estivesse diminuindo devido à redução do fluxo do rio, eles teriam migrado para uma fonte de água mais constante", disse Nick Scroxton
, pesquisador de hidrologia, paleoclima e paleoambientes do University
College Dublin e coautor do artigo, em um e-mail para o Live Science.
"Portanto, faz sentido que os hobbits os tenham seguido."
Reconstrução do Homo floresiensis . (Crédito da imagem: Foto de Bill O'Leary/The Washington Post. via Getty Images)
É possível que a mudança para o litoral tenha colocado os hobbits em contato com grupos de Homo sapiens
que estavam se expandindo pela região. Esse contato poderia ter
resultado em competição por recursos e até mesmo em conflitos entre os
grupos, sugeriu Scroxton. Além disso, a erupção vulcânica de cerca de
50.000 anos atrás teria piorado ainda mais a situação para os hobbits.
"Este parece ser um estudo muito impressionante", disse Julien Luoys
, paleontólogo da Universidade Griffith, na Austrália, que realizou
extensas pesquisas sobre hominídeos, mas não participou da nova
pesquisa, em um e-mail para a Live Science. Ele observou que uma redução
nas chuvas pode ter um grande impacto em uma ilha tão pequena quanto
Flores.
"Há
uma quantidade limitada de espaço em uma ilha, e apenas um número
limitado de tipos de ambientes que podem ser abrigados", disse Luoys.
"Quando as coisas ficam mais secas, um animal não pode simplesmente sair
da ilha, e quaisquer refúgios potenciais que ele pudesse usar irão
desaparecer ou ficar muito lotados, muito rapidamente."
Debbie Argue
, professora honorária da Escola de Arqueologia e Antropologia da
Universidade Nacional da Austrália, que não participou do estudo, também
elogiou a pesquisa. "O artigo nos oferece uma excelente visão sobre as
mudanças climáticas na região e é uma contribuição muito bem-vinda para o
conhecimento sobre as condições passadas em Flores", disse Argue ao
Live Science por e-mail.
Explorando o comportamento humano por meio de análises isotópicas: ferramentas, escalas e questões.
Capítulo
Primeiro online:
Resumo
As
análises isotópicas tornaram-se uma ferramenta importante na pesquisa
arqueológica e bioarqueológica. Este capítulo apresenta uma visão geral
da aplicação de análises isotópicas, tanto estáveis quanto
radiogênicas, na reconstrução do comportamento humano no passado.
Apresentamos uma visão geral dos principais sistemas isotópicos
comumente usados nesse contexto, ou seja, carbono, nitrogênio,
enxofre, oxigênio e estrôncio. Em seguida, discutimos os tecidos mais
comuns dos quais as razões isotópicas são obtidas e destacamos a
importância de se considerar as escalas de análise no planejamento da
pesquisa e na interpretação dos dados. Finalmente, apresentamos as
categorias gerais de questões de pesquisa social para as quais os
estudos isotópicos têm sido particularmente produtivos, como estudos de
paleodieta e história de vida, interações humano-animal e mobilidade e
migração humana. Dentro dessas categorias gerais, discutimos as
contribuições individuais para o tema.
Desde
a década de 1950, os avanços na pesquisa isotópica revolucionaram
estudos em diversas áreas, incluindo geologia, ecologia e biologia. No
âmbito das ciências sociais, a arqueologia e a bioarqueologia se
beneficiaram particularmente do desenvolvimento e da adoção desses novos
métodos analíticos, obtendo novas vias de investigação sobre o
comportamento humano no passado. Ao compreender como as proporções
isotópicas se alteram à medida que circulam em sistemas biológicos,
hidrológicos e geológicos, muitos aspectos do comportamento humano
habitual podem ser avaliados e quantificados por meio de análises de
tecidos biológicos preservados. Essas análises são agora rotineiramente
empregadas em pesquisas arqueológicas e bioarqueológicas para explorar
uma gama de questões em diferentes escalas de análise. Este volume reúne
pesquisadores que trabalham com diferentes sistemas isotópicos e
destaca as diversas maneiras pelas quais esses métodos podem ser
utilizados para reconstruir aspectos do comportamento humano no passado.
As ações humanas são influenciadas por circunstâncias locais e
imediatas, ao mesmo tempo que são incentivadas ou restringidas por
fatores culturais e ambientais mais amplos. Por essa razão, pesquisas
que utilizam análises isotópicas no estudo do comportamento humano
também devem levar em consideração as escalas espaciais e temporais em
que suas questões são mais relevantes. Análises isotópicas de diferentes
tecidos corporais refletem diferentes escalas temporais, dependendo das
taxas de renovação e dos processos de desenvolvimento tecidual. O
tamanho da amostra analisada e a extensão geográfica de onde ela é
extraída, além disso, têm implicações significativas para a escala de
análise em consideração. Uma compreensão mais completa do comportamento
humano no passado pode ser obtida considerando-se a dinâmica em
múltiplas escalas de análise e avaliando como os comportamentos em uma
escala interagem e influenciam comportamentos em escalas mais amplas ou
mais específicas (por exemplo, Zvelebil & Weber, 2013 ).
O
objetivo deste volume é fornecer revisões e estudos de caso de
especialistas na aplicação de métodos isotópicos a questões do
comportamento humano em diferentes escalas temporais e espaciais. Este
volume destaca a utilidade de vários isótopos (δ) 13 Cd 15 N, d 34 S, d 18 O, e 87 Sr./ 86 Sr)
em tais estudos. O foco deste volume não está nos elementos em si, mas
sim nas questões sociais que eles nos permitem abordar. Como disciplina
que se concentra em padrões e processos de longo prazo do comportamento
humano, a arqueologia está em uma posição única para contribuir com
pesquisas sobre alguns dos grandes desafios enfrentados pelas sociedades
humanas modernas, incluindo as adaptações humanas às mudanças
climáticas, a desigualdade socioeconômica e as migrações humanas em
larga escala. As análises isotópicas, quando combinadas com questões e
hipóteses teoricamente fundamentadas, são capazes de fornecer dados
quantitativos de sociedades passadas que são relevantes para muitas
questões contemporâneas.
Neste capítulo, apresentamos uma
estrutura para incorporar estudos isotópicos em pesquisas que exploram o
comportamento humano no passado. Em vez de fornecer uma revisão
completa dos princípios básicos da análise isotópica (como já foi feito
por outros autores, por exemplo, Fry, 2007 ; Katzenberg & Waters-Rist, 2018
), oferecemos uma breve introdução para este volume, apresentando os
elementos mais comuns utilizados em pesquisas arqueológicas e
bioarqueológicas e discutindo considerações fundamentais em estudos
isotópicos, incluindo os tipos de tecido analisados, bem como questões
de diagênese, precisão e exatidão. Também enfatizamos a importância de
reconhecer as escalas de análise como um fator crucial ao aplicar
métodos isotópicos a questões de comportamento humano. Por fim,
destacamos os estudos de caso e revisões presentes neste volume,
pertinentes às categorias gerais de questões de pesquisa social para as
quais os estudos isotópicos têm se mostrado particularmente produtivos:
(1) paleodieta e história de vida, (2) interações humano-animal e (3)
mobilidade e migração humana.
2.2 Elementos de Interesse
Isótopos
são variantes de elementos químicos que possuem o mesmo número de
prótons e elétrons, mas diferem no número de nêutrons. A utilidade das
análises isotópicas em investigações ecológicas reside no fato de que as
proporções de diferentes isótopos do mesmo elemento variam de acordo
com padrões previsíveis em sistemas biológicos, hidrológicos e
geológicos. Padrões ou diferenças observados nessas proporções revelam
informações sobre o comportamento, a localização e a história dos
organismos dentro desses contextos mais amplos. Isótopos estáveis, como
carbono, nitrogênio, enxofre e oxigênio, são expressos pela notação
delta (δ), que reflete a variação na proporção do isótopo raro em
relação ao isótopo abundante, comparativamente a um padrão
internacional, e são relatados em partes por mil pelo símbolo ‰
(permil). Para o estrôncio, a abundância relativa de isótopos
radiogênicos é representada pela notação delta (δ). 87 O Sr é normalizado em relação ao não radiogênico. 86 Senhor, apresentado como 87 Sr./ 86 Senhor,
portanto, a notação delta e o símbolo permil não são usados para
reportar valores. Esses cinco sistemas isotópicos são brevemente
apresentados e revisados abaixo.
2.2.1 Princípios da Análise de Isótopos Estáveis de Carbono
A proporção dos dois principais isótopos estáveis de carbono ( 13 C/ 12 C) é expresso em relação a um padrão internacional (Vienna Pee Dee Belemnite; VPDB) como δ 13 C.
Este padrão foi originalmente composto por uma amostra de fóssil de
belemnite do Cretáceo encontrada na formação calcária Pee Dee (PDB) na
Carolina do Sul, mas foi esgotado pelo uso. Um novo VPDB tornou-se o
padrão internacional substituto com base na referência (+1,95‰) ao
original (Coplen et al., 2006
). Substituições semelhantes de padrões internacionais ocorreram para
outros isótopos (ou seja, VSMOW para oxigênio e VCDT para enxofre). A
principal fonte de variação em δ 13 Os valores de C em sistemas biológicos representam as diferentes vias fotossintéticas das plantas (DeNiro & Epstein, 1978 ; O'Leary, 1988 ; Smith & Epstein, 1971 ). A maioria das plantas utiliza a via fotossintética C3 ( Calvin-Benson) e apresenta δ 13 Valores de C entre −35 e −20 por mil (‰) (Kohn, 2010 ). Plantas adaptadas a ambientes áridos, particularmente gramíneas tropicais como o milho ( Zea mays ), utilizam a via fotossintética C4 ( Hatch-Slack), que resulta em valores relativamente mais altos (menos negativos), tipicamente entre −15 e −7‰ (O'Leary, 1988
). O Metabolismo Ácido das Crassuláceas (CAM) é um terceiro tipo de
fotossíntese utilizado por suculentas, bromélias e orquídeas. Plantas
CAM utilizam aspectos das fotossínteses C3 e C4 e podem apresentar δ 13 Valores de C que abrangem toda a gama de plantas C3 a C4 ( Kluge & Ting, 1978 ). Plantas CAM de ambientes xéricos, como Cactaceae e Agavaceae, exibem . mais elevado ( semelhante a C4) δ 13 valores de C do que plantas CAM de regiões mais úmidas, que exibem mais baixo (tipo C3 ) δ 13 Valores C (Borland et al., 2011 ; Winter & Smith, 1996 ).
As proporções relativas de plantas C3 , C4 ou CAM na dieta de um organismo refletem-se no δ medido. 13 Os valores de C dos tecidos do consumidor . são maiores em organismos que consomem plantas C4 do que em organismos que consomem plantas C3 Em ecossistemas costeiros e estuarinos, os organismos incorporam carbono do bicarbonato dissolvido que possui δ 13 Valores de C que se aproximam dos de plantas C4 ( Hemminga & Mateo, 1996 ; Schwarcz & Schoeninger, 1991 ). Alguns plânctons marinhos apresentam valores intermediários entre plantas C3 e C4 ,
enquanto plantas aquáticas de água doce obtêm carbono do carbono
inorgânico dissolvido em águas naturais e possuem valores que se
sobrepõem aos de plantas C3 ( Schwarcz & Schoeninger, 1991 ).
Diferentes tecidos incorporam o carbono da dieta de acordo com princípios diferentes. O carbonato mineral ósseo (
), por exemplo, deriva carbono de todas as fontes alimentares – incluindo proteínas, carboidratos e lipídios – e, portanto, o δ do carbonato 13 Os valores de C refletem o δ 13 C da dieta total (Ambrose & Norr, 1993 ; Froehle et al., 2010 ; Krueger & Sullivan, 1984 ; Lee-Thorp et al., 1989 ; Schwarcz, 2000 ). Colágeno ósseo δ 13 Os
valores de C, por outro lado, são tendenciosos em relação às fontes de
proteína alimentar, visto que cerca de 75% dos átomos de carbono no
colágeno se originam de proteínas alimentares (Fernandes et al., 2012
). Análises de qualquer uma dessas fases ósseas, ou de ambas em
conjunto, revelam informações sobre as práticas alimentares de
organismos do passado (Froehle et al., 2010
) e possivelmente sobre a mobilidade quando a disponibilidade de
recursos alimentares varia de acordo com a região (por exemplo,
Somerville et al., 2020 ).
2.2.2 Princípios da Análise de Isótopos Estáveis de Nitrogênio
A proporção dos dois principais isótopos estáveis de nitrogênio ( 15 N/ 14 N) é expresso em notação delta em relação ao padrão internacional de N 2 atmosférico (AIR) como δ 15 N. Para os herbívoros, a principal fonte de nitrogênio provém das plantas em sua dieta (DeNiro & Epstein, 1981 ). Espécies de plantas leguminosas tipicamente exibem δ 15 Os
valores de N próximos de 0‰ são obtidos porque eles adquirem grande
parte do seu nitrogênio fixando diretamente o N₂ atmosférico , que por definição tem um δ 15 O valor de N é de 0‰ (ou seja, o padrão internacional) (Virginia & Delwiche, 1982 ). A maioria das outras plantas obtém nitrogênio de nitratos do solo (
, amônio do solo
e aminoácidos do solo (Robinson, 2001 ), e são enriquecidas em vários permil em relação ao δ atmosférico. 15 N. Um aumento gradual em δ 15 Valores
de N de 3–5‰ ocorrem com posições ascendentes em um sistema trófico, de
modo que herbívoros exibem valores mais altos do que plantas, onívoros
exibem valores mais altos do que herbívoros e carnívoros exibem valores
mais altos do que onívoros (Minagawa & Wada, 1984 ; Schoeninger & DeNiro, 1984 ). Devido a esse processo de enriquecimento trófico em 15 N, estudos arqueológicos frequentemente usam δ 15 Os
valores N dos tecidos do consumidor são utilizados para quantificar a
quantidade de carnivoria na dieta (ou seja, posição trófica) ou os tipos
de fontes de proteína que foram consumidas regularmente (DeNiro &
Epstein, 1981 ; Hedges & Reynard, 2007 ; Schoeninger & DeNiro, 1984 ).
Organismos de ambientes marinhos tendem a apresentar δ mais elevado. 15 Os
valores de N são maiores do que os de organismos terrestres devido ao
maior número de etapas tróficas nos ecossistemas marinhos e ao δ
geralmente elevado. 15 Valores de N do nitrato e da matéria orgânica particulada no oceano (Fry, 2007 ; Guo et al., 2004 ; Schoeninger et al., 1983 ). Assim, para populações arqueológicas que consumiam uma mistura de recursos terrestres e marinhos, δ 15 Os
valores N dos consumidores podem elucidar a dependência relativa de
diferentes fontes de proteína, particularmente quando usados em
combinação com δ. 13 Valores C.
Outras considerações relativas à interpretação de δ 15 Os
valores de N incluem a influência de fertilizantes artificiais,
influências ambientais e fisiologia. Estudos experimentais e de campo
demonstraram que práticas agrícolas antigas, incluindo a fertilização,
podem causar valores artificialmente mais altos em plantas e nos animais
que as consomem, complicando a interpretação do comportamento humano no
passado (Szpak et al., 2012 ; veja também a discussão em Kinaston, neste volume). Além disso, o δ das plantas 15 Os
valores de N são influenciados por aspectos do ambiente local, como a
aridez e a salinidade do solo em que foram cultivados (Hartman, 2011 ; Ugan & Coltrain, 2011 ). δ da planta 15 Os valores de N tendem a ser mais altos em regiões quentes e secas e mais baixos em regiões frias e úmidas porque o δ 15 Os
valores de N dos nitratos e da amônia no solo são influenciados por
variáveis ambientais como temperatura, umidade e atividade microbiana
(Amundson et al., 2003 ; Craine et al., 2015a , b ). Em tecidos animais, fatores adicionais influenciam o δ medido. 15 Os
valores de N incluem doença, inanição, crescimento e gravidez devido à
sua influência no balanço de nitrogênio no organismo (por exemplo,
Fuller et al., 2005 ). O estado fisiológico e metabólico deve, portanto, ser levado em consideração ao interpretar δ. 15 Valores N (Katzenberg & Waters-Rist, 2018 ; Reitsema, 2013 ).
2.2.3 Princípios da Análise de Isótopos Estáveis de Enxofre
A proporção dos dois isótopos estáveis mais abundantes de enxofre ( 34 S/ 32 S) é apresentado em relação ao padrão internacional Vienna Canyon Diablo Troilite (VCDT) como δ 34 O
enxofre está naturalmente presente na atmosfera, geosfera, hidrosfera e
biosfera. As plantas adquirem enxofre principalmente do dióxido de
enxofre atmosférico (SO₂ ) , do sulfato do solo
e do SO₄²⁻ dissolvido
em ambientes aquáticos, assimilando-o nos aminoácidos cisteína e metionina (Agrawal, 2003 ; Krouse & Tabatabai, 1986
. Os consumidores de plantas, por sua vez, assimilam o enxofre dos
aminoácidos vegetais em seus tecidos. Humanos e outros onívoros adquirem
enxofre das proteínas de sua dieta, que podem ser de origem vegetal ou
animal. Efeitos tróficos altamente variáveis, mas geralmente pequenos,
têm sido observados em diferentes tecidos, organismos e dietas, o que
complica o uso de δ 34 S como indicador de posição trófica (Nehlich, 2015
). Pesquisas experimentais recentes sugerem um desvio geral de cerca de
−1,5‰ entre a dieta e os tecidos do consumidor (Webb et al., 2017 ), embora isso não seja observado de forma consistente em estudos de campo (Krajcarz et al., 2019 ).
A
análise de isótopos estáveis de enxofre em arqueologia tem sido
aplicada com sucesso em estudos de paleodieta e mobilidade. Como as
proporções de isótopos de enxofre em plantas são bastante diferentes
entre ambientes marinhos, de água doce e terrestres, a análise de
isótopos estáveis de enxofre em arqueologia tem se mostrado útil para
estimar as contribuições proporcionais de alimentos de diferentes fontes
de recursos, particularmente diferenciando entre o consumo de peixes
marinhos ou de água doce (Nehlich et al., 2010 ; Richards et al., 2001 ). Regionalmente, δ 34 Os
valores de S variam nos solos e plantas locais devido a fatores como a
distância do mar e a natureza da geologia local (Kabalika et al., 2020
). Essa heterogeneidade geográfica permite o uso da análise de isótopos
estáveis de enxofre como ferramenta para rastrear mudanças
residenciais e migrações passadas (Ebert et al., 2021 ; Vika, 2009 ).
2.2.4 Princípios da Análise de Isótopos Estáveis de Oxigênio
A proporção dos dois principais isótopos estáveis de oxigênio ( 18 O/ 16 O) é apresentado em relação a um padrão internacional (VPDB ou Standard Mean Ocean Water; SMOW) como δ 18 O
oxigênio encontrado na apatita, um mineral ósseo, deriva, em última
análise, do oxigênio da água consumida, que pode ser água proveniente da
alimentação (como a água das folhas), água superficial ingerida ou água
obtida pela respiração (Kohn et al., 1996 ; Longinelli, 1984 ; Luz et al., 1984
). Organismos que não são bebedores obrigatórios, como cangurus ou
coelhos, obtêm água principalmente por meio do consumo de material
vegetal e apresentam valores estáveis de isótopos de oxigênio ( δ). 18 O apatita ) que se correlacionam negativamente com a umidade relativa (Ayliffe & Chivas, 1990 ; Levin et al., 2006 ) e com a precipitação média anual (Somerville et al., 2018 ). Esse processo se deve à evaporação preferencial de 16 O proveniente das folhas em locais áridos ou em estações mais secas, levando a uma maior concentração de oxigênio. 18 O/ 16 Razões de O na água das folhas. Animais que obtêm a maior parte da sua água bebendo água superficial, como os humanos, exibem δ 18 Os valores de apatita que apresentam maior correlação com as temperaturas locais são δ 18 Os valores de δ¹⁸O da água meteórica são fortemente influenciados por variações de temperatura (Fricke & O'Neil, 1999 ; Hallin et al., 2012 ; Longinelli, 1984 ; Luz et al., 1990 ). Entre regiões, a altitude e a distância do mar também atuam como fatores importantes na variação de δ¹⁸O. 18 Os valores de O nas fontes de água e, consequentemente, nos organismos que as consomem. Essa variação permitiu o uso de δ 18 Os
valores de O são um meio de rastrear a mobilidade geográfica de humanos
e outros organismos na paisagem, além de fornecer informações sobre o
paleoambiente (Bowen et al., 2005 ; Killingley & Lutcavage, 1983 ; White et al., 2000 ).
Assim como os valores de isótopos estáveis de carbono, δ 18 Os valores de isótopos de oxigênio da apatita
podem ser obtidos a partir de diferentes componentes do tecido ósseo.
Os valores de isótopos de oxigênio podem ser obtidos dos grupos fosfato (
) ou carbonato ( ) do mineral hidroxiapatita do osso e do esmalte. Os valores de isótopos estáveis de oxigênio da apatita e
não são imediatamente equivalentes, mas apresentam correlação linear,
uma vez que ambos estão em equilíbrio com a água corporal e, portanto,
podem ser convertidos um no outro de acordo com fórmulas padrão (Iacumin
et al., 1996 ).
2.2.5 Princípios da Análise de Isótopos de Estrôncio Radiogênico
O
estrôncio é um elemento natural encontrado na crosta terrestre e está
presente em rochas, solos e água. Possui três isótopos estáveis não
radiogênicos ( 84 Senhor, 86 Senhor, e 88 Sr) e um isótopo radiogênico estável, 87 Sr, que é o produto do decaimento do rubídio-87 radioativo. Em arqueologia e bioarqueologia, estudos sobre a variação de 87 Sr./ 86 As proporções de Sr em tecidos biológicos podem fornecer informações sobre migração ou mobilidade residencial (Ericson, 1985 ; Price et al., 2008 ; Wright, 2005 ). Tais estudos baseiam-se na observação de que 87 Sr./ 86 As
proporções de Sr nos ossos e dentes são derivadas, em última análise,
do estrôncio nos substratos do solo, e essas proporções variam
significativamente entre as regiões (Capo et al., 1998 ; Faure & Powell, 1972 ; Hodell et al., 2004 ).
Os fatores que influenciam a variação regional em 87 Sr./ 86 O Sr inclui as proporções de estrôncio e rubídio na rocha matriz (Rb/Sr), o original 87 Sr./ 86 O valor de Sr da rocha no momento da cristalização e a idade da rocha matriz (Bentley, 2006
) são fatores importantes. Geralmente, espera-se que materiais
geológicos mais antigos, com altas razões Rb/Sr naturais, apresentem os
valores mais elevados. 87 Sr./ 86 O Sr, enquanto materiais geológicos mais jovens com menores proporções de Rb/Sr devem apresentar a menor concentração. 87 Sr./ 86 Entre
os diferentes tipos de rocha matriz, arenitos e granitos com baixo teor
de cálcio tendem a apresentar a maior razão Rb/Sr, enquanto carbonatos e
basaltos apresentam razões Rb/Sr muito menores (Bentley, 2006
: 140). O intemperismo das rochas libera estrôncio no solo e nas águas
subterrâneas, e este entra nos sistemas biológicos por meio de sua
incorporação em material vegetal (Burger & Lichtscheidl, 2019 ; Faure & Mensing, 2005 ; Sillen et al., 1998 ).
Os
herbívoros adquirem estrôncio das plantas que consomem, enquanto os
carnívoros o adquirem indiretamente ao se alimentarem de herbívoros. O
estrôncio é incorporado ao tecido ósseo e ao esmalte como Sr. 2+ substitutos para Ca 2+ dentro da hidroxiapatita devido à sua configuração eletrônica semelhante (Comar et al., 1957 ; Ezzo, 1994
). O fracionamento dependente da massa das razões isotópicas do
estrôncio ocorre devido a processos naturais e a práticas de tratamento e
análise em laboratório (por exemplo, Neymark et al., 2014 ), mas a prática de normalizar as razões isotópicas do estrôncio a uma constante 86 Sr./ 88 A razão Sr (0,1194) permite aos pesquisadores avaliar a variação em 87 Sr./ 86 razões de Sr devido à variação natural no substrato geológico (Nier, 1938 ). Devido a essas correções e à diferença de massa relativa entre 87 Senhor e 86 O Sr é muito menor do que os das variáveis de isótopos estáveis mais leves (δ 13 Cd 15 N, d 34 S, d 18 O), espera-se que plantas, herbívoros e predadores exibam comportamentos semelhantes. 87 Sr./ 86 Sr dentro de uma determinada região. Portanto, comparando 87 Sr./ 86 A
análise de Sr entre diferentes indivíduos, ou entre indivíduos e a
geologia local, ou mesmo entre o osso e o esmalte de um indivíduo,
permite-nos observar a mobilidade dos organismos entre diferentes
regiões geológicas (Borić & Price, 2013 ; Knudson et al., 2004 ; Price et al., 2002 ; Schwartz et al., 2021 ; Thornton, 2011 ).
2.2.6 Outros isótopos de interesse antropológico
Além
da lista acima de traçadores isotópicos, novas pesquisas estão
demonstrando a utilidade de vários outros sistemas isotópicos para
abordar questões sobre o comportamento humano. Embora não apresentemos
aqui estudos de caso ou revisões desses sistemas, observamos que
pesquisas focadas em isótopos de hidrogênio (France et al., 2018 ; Reynard & Hedges, 2008 ), chumbo (Sharpe et al., 2016 ; Turner et al., 2009 ), zinco (Jaouen et al., 2016 ; McCormack et al., 2021 ), cobre (Jaouen et al., 2017 ) e cálcio (Reynard et al., 2011
) provavelmente ampliarão a lista de questões de pesquisa às quais
pesquisadores que investigam o comportamento humano no passado podem
aplicar análises isotópicas.
2.3 Considerações para Estudos Isotópicos
2.3.1 Tipos de tecido analisados
As
razões isotópicas estáveis e radiogênicas podem ser obtidas tanto de
tecidos orgânicos quanto inorgânicos. Em aplicações antropológicas de
análises de isótopos estáveis, o tecido biológico mais comumente
analisado em estudos do comportamento humano no passado é o colágeno
ósseo. Embora a fase orgânica do osso seja comumente referida como
"colágeno", a menos que a composição de aminoácidos do material tenha
sido determinada, o que os pesquisadores denominam "colágeno" na
literatura tipicamente contém cerca de 10% de materiais orgânicos não
colagenosos em peso em mamíferos (Fisher et al., 1987 ; Schwarcz & Schoeninger, 1991
). A dentina dentária, assim como o osso, também pode ser analisada
quanto às razões isotópicas do componente orgânico colágeno (Beaumont
& Montgomery, 2016 ; Eerkens et al., 2011
). Valores de carbono, nitrogênio e enxofre estáveis também são
rotineiramente analisados em tecidos orgânicos compostos de queratina,
como cabelo e unhas. Embora cabelos e unhas não se preservem por tanto
tempo no registro arqueológico, estudos em ambientes frios (Britton et
al., 2018 ; Macko et al., 1993 Em ambientes secos (White, ) , ; White et al., 2009
esses tecidos queratinosos têm sido utilizados devido às condições
favoráveis à sua preservação. Quanto aos tecidos inorgânicos
(minerais), o osso e o esmalte têm sido os mais comumente analisados,
particularmente em projetos que utilizam valores isotópicos de
estrôncio, oxigênio e, mais recentemente, chumbo para explorar a
paleomobilidade e o paleoambiente. Os valores isotópicos de diferentes
tecidos são produto de processos variáveis de desenvolvimento e
renovação, com diferentes tecidos representando diferentes períodos da
vida de um indivíduo. O momento do desenvolvimento e a taxa de renovação
do tecido selecionado devem ser levados em consideração durante o
planejamento de qualquer projeto de pesquisa para garantir que uma
amostra apropriada esteja sendo utilizada. Essa questão da escala de
análise com base no tecido selecionado é discutida com mais detalhes a
seguir.
2.3.2 Diagênese
Embora
os primeiros estudos reconhecessem que as proporções isotópicas dos
tecidos de um organismo registram aspectos de sua dieta e ecologia em
vida, foi somente quando os pesquisadores conseguiram caracterizar a
alteração diagenética dos ossos e demonstrar a preservação de valores
biogênicos que a pesquisa isotópica se tornou rotina na pesquisa
arqueológica (Vaiglova et al., 2022
). Atualmente, os métodos de controle de qualidade mais comuns para
garantir dados confiáveis de materiais orgânicos, como o colágeno,
incluem avaliações das proporções C:N (DeNiro, 1985 ; Guiry & Szpak, 2021
), com uma faixa aceitável de 2,9 a 3,6, e o percentual de rendimento
de colágeno ósseo após a desmineralização, partindo-se do pressuposto de
que um rendimento inferior a 1% resulta em dados não confiáveis
(Ambrose, 1990
). Para tecidos mineralizados inorgânicos, as técnicas comuns para
triagem de diagênese incluem espectroscopia de infravermelho com
transformada de Fourier (FTIR) (Beasley et al., 2014 ; France et al., 2020 ; Weiner & Bar-Yosef, 1990 ; Wright & Schwarcz, 1996 ) e rendimento de bioapatita (Chesson et al., 2021). ), e a análise das concentrações de elementos traço (Kohn et al., 1999 ; Trueman & Tuross, 2002
). Para FTIR, as medidas comuns incluem o fator de divisão
infravermelha (IR-SF, também conhecido como índice de cristalinidade),
com faixas aceitáveis variando de acordo com o tipo de método
utilizado, e a razão carbonato/fosfato (C:P), com faixas aceitáveis
variando entre 0,1 e 0,4 (Beasley et al., 2014
). Para o rendimento de bioapatita, uma porcentagem em peso entre 21 e
63% de apatita remanescente após o tratamento da amostra é considerada
aceitável (Chesson et al., 2021
). As análises elementares de osso e esmalte partem do pressuposto de
que a presença de elementos raros não encontrados em osso fresco, como
urânio ou lantânio, indicaria absorção post-mortem e, portanto,
diagênese (Kohn et al., 1999 ; Trueman & Tuross, 2002 ).
2.3.3 Precisão e Exatidão
Além
de garantir valores isotópicos biogênicos, a precisão e a exatidão
analíticas dos dados produzidos pela espectrometria de massa são
essenciais. Diversas revisões abordam as melhores práticas para
assegurar medições isotópicas confiáveis e os meios mais adequados
para relatar os dados (ver Szpak et al., 2017 ; Vaiglova et al., 2022
). Estudos recentes destacam a variabilidade existente entre
laboratórios em relação aos métodos de preparação, instrumentação e
procedimentos de calibração (Chesson et al., 2021 ; Edwards et al., 2022 ; Pestle et al., 2014
). Essas diferenças interlaboratoriais podem resultar em valores
isotópicos medidos significativamente diferentes, que acumulam erros
adicionais além da variação natural nos valores isotópicos que existe
dentro dos organismos (Berg et al., 2022
). Compreender essas fontes de variação é crucial ao se extrair
conclusões comportamentais a partir de dados isotópicos (Tabela 2.1 ). Pestle e colegas ( 2014
) forneceram a 21 laboratórios ao redor do mundo 5 subamostras de um
fêmur arqueológico para preparação e análise de isótopos de carbono e
nitrogênio do colágeno ósseo e carbono e oxigênio da bioapatita óssea. A
variação resultante nos valores foi suficientemente grande para levar a
diferenças interpretativas no comportamento humano no passado,
particularmente para os dados de isótopos estáveis de oxigênio. Esses
resultados servem de alerta e devem levar todos os profissionais a
refletir: quão estáveis, biogênicos e comparáveis são os valores
resultantes das análises isotópicas? Foi proposto que uma Diferença
Mínima Significativa (DMS) seja utilizada como o limiar estabelecido
para quando duas análises devem ser consideradas como apresentando uma
diferença real em termos de comportamento, em contraste com a mera
variabilidade interlaboratorial (Pestle et al., 2014). 2021 Chesson e colegas ( 2021
) propuseram que, além da MMD, uma Diferença Interpretativa Real (DIR)
fosse considerada como um limiar adicional de diferença significativa,
por meio da preparação de 30 amostras idênticas em dois laboratórios
distintos, seguindo o mesmo protocolo e sendo analisadas no mesmo
laboratório de instrumentação (Chesson et al., ; Edwards et al., 2022 ). Berg et al. ( 2022
) analisaram a variação intraindividual nos valores isotópicos,
examinando 5 ou 6 ossos longos de 27 humanos modernos para quantificar o
limiar de variação intraorganismo que deveria ser considerado para
humanos. A Tabela 2.1
destaca a importância de se considerar a precisão e a exatidão dos
valores gerados por análises isotópicas antes que pesquisadores da área
de arqueologia proponham interpretações sobre o comportamento humano no
passado.
Tabela
2.1 Comparação da variação interlaboratorial e intraindividual de
interesse para pesquisadores em arqueologia (todos os valores expressos
em ‰)
Desde
a década de 1970, as análises isotópicas evoluíram de processos de
extração trabalhosos para análise de elementos individuais em gasodutos
para amostragem automatizada que combina análises de múltiplos elementos
a partir de uma única preparação de amostra. Com o aprimoramento dos
métodos de espectrometria de massa de razão isotópica (IRMS), também
houve avanços na interpretação dos isótopos. Os primeiros estudos
frequentemente consistiam apenas em análises de elementos individuais,
destacando diferenças entre espécies para compreender as compensações
dietéticas e a posição trófica dentro de uma teia alimentar (Chisholm et
al., 1982
). Gráficos bivariados de isótopos analisados em conjunto ainda são
as representações visuais mais comuns dos valores isotópicos, auxiliando
na compreensão dos dados e fundamentando as interpretações.
O
início dos anos 2000 trouxe uma onda de novas metodologias de outras
disciplinas, como ecologia e geoquímica, relevantes para a interpretação
do comportamento humano. Essas novas abordagens incluíram modelos
formais de mistura (Phillips & Koch, 2002 ), estatística bayesiana (Semmens et al., 2009 ) e mapas de isótopos (Bowen, 2010
). Os modelos de mistura foram introduzidos aos arqueólogos como uma
forma de quantificar e compreender as contribuições proporcionais de
diferentes fontes alimentares na dieta de populações antigas (Phillips
& Koch, 2002
). Desde então, os modelos de mistura variaram de modelos lineares a
modelos probabilísticos simples e abordagens bayesianas mais
sofisticadas (Cheung & Szpak, 2021
). Por definição, os modelos são simplificações da realidade e todos
apresentam vantagens e desvantagens que os arqueólogos devem considerar
ao selecionar uma abordagem para interpretar seus dados (ver revisão de
Cheung & Szpak, 2021
). Foi demonstrado que os isótopos mudam sistematicamente ao longo do
espaço geográfico, como uma paisagem isotópica, portanto, quando os
valores isotópicos são modelados com sistemas de informação geográfica
(SIG), resulta em uma paisagem isotópica interpretativa (Bowen et al., (2005
). O mapa isotópico resultante pode servir como um "instantâneo" para
auxiliar na interpretação dos valores isotópicos, permitindo estudos
detalhados sobre história de vida, migração, distribuição de recursos
alimentares ou mudanças nas condições ambientais. Novas formas de
modelar sistemas isotópicos múltiplos como funções discriminantes também
podem apoiar novas interpretações do comportamento humano no passado.
Os arqueólogos devem considerar a melhor forma de analisar e interpretar
seus dados com essas abordagens variadas, dependendo das questões de
pesquisa.
2.4 Escalas de Análise
A
complexidade do comportamento humano exige considerações em diferentes
escalas de análise, particularmente quando se tenta compreender padrões
de comportamento antigos e de longo prazo por meio de análises
isotópicas. As escalas temporais e geográficas variam em ordens de
magnitude em relação ao alcance do comportamento humano (Tabela 2.2
). Como agentes capazes de respostas criativas e adaptativas a
circunstâncias em constante mudança, os humanos frequentemente tomam
decisões sobre ações apropriadas em um local específico na escala de
segundos a minutos. No entanto, esses comportamentos são frequentemente
restringidos, incentivados ou estruturados por padrões sociais de maior
escala, como papéis de gênero, estruturas de classe ou costumes e
tradições culturais (por exemplo, Bourdieu, 1977 ; Giddens, 1979 ; Sewell, 1992 ), que são melhor compreendidos em regiões mais amplas em resoluções temporais que operam em escalas de anos a séculos.
Tabela 2.2 Escalas de análise relevantes para estudos do comportamento humano por meio de métodos isotópicos
Como
demonstramos neste livro, os métodos isotópicos são capazes de
investigar o comportamento em escalas que podem variar de vários dias a
semanas, anos e séculos. Os processos sociais na escala de dias a
semanas incluem eventos como festas, mudanças sazonais na dieta e
mudanças recentes no local de residência. Ao se concentrarem nessa
resolução de escala fina, os pesquisadores se preocupam principalmente
com indivíduos ou pequenos grupos de indivíduos e suas histórias de vida
individuais. Como o cabelo, as unhas e o esmalte são inertes após a
formação, eles representam as contribuições isotópicas no momento da
síntese inicial do tecido e, portanto, são os melhores tecidos para
amostrar ao investigar questões nessa escala de tempo. Com base em
estudos anteriores, podemos fazer as seguintes generalizações sobre a
taxa de crescimento de diferentes tecidos, embora todas as taxas possam
variar de acordo com a idade, o sexo e o estado de saúde. O cabelo
humano tende a crescer a uma taxa de 1,05 cm/mês (Saitoh et al., 1970
). As unhas das mãos crescem a uma taxa de 0,35 mm/mês, enquanto as
unhas dos pés crescem a uma taxa de 0,16 cm/mês (Yaemsiri et al., 2010
). Os dentes se desenvolvem em diferentes idades ao longo da vida de um
indivíduo e variam de acordo com o tipo de dente, mas, geralmente, as
coroas dos dentes mineralizam ao longo de aproximadamente 3 a 6 anos e
as raízes mineralizam ao longo de vários anos adicionais (Hägg &
Matsson, 1985 ; Hillson, 1996
). Isso significa que, dependendo do comprimento ou tamanho da amostra,
uma amostra seriada de cabelo pode refletir um período de um mês a um
ano antes da morte, uma unha pode representar um período de cinco a seis
meses antes da morte, enquanto um dente refletiria valores registrados
durante a infância. A amostragem intra-tecidual de cabelo, unhas e
dentes pode, portanto, registrar mudanças isotópicas em curtos
intervalos de tempo em diferentes fases da história de vida e fornecer
informações de alta resolução sobre atividades humanas passadas.
Os
tipos de comportamentos humanos que operam na escala de semanas a meses
incluem comportamentos mais padronizados, como a produção sazonal de
alimentos, rotas migratórias anuais ou calendários rituais. Como as
sociedades humanas frequentemente se adaptam ou são influenciadas por
flutuações sazonais de recursos, as tradições culturais e as estratégias
de obtenção de alimentos geralmente mudam ao longo do ano. De fato,
estruturas e normas sociais complexas podem ser contingentes
sazonalmente e algumas sociedades, como a dos Inuit, oscilam entre
arranjos sociais hierárquicos e igualitários ao longo do ano (Graeber
& Wengrow, 2021 ; Mauss, 2013
). Análises isotópicas podem abordar a variação nos padrões de dieta e
mobilidade nessa escala por meio do uso de estratégias de amostragem
intra-cabelo, cabelo em massa, intra-unha, unha em massa, intra-esmalte,
intra-dentina e intra-osso.
Padrões semelhantes também podem ser
avaliados em uma escala ligeiramente mais ampla, de vários meses a
anos, incluindo comportamentos similares, como mudança de residência e
padrões de práticas alimentares que ocorrem em nível de locais,
assentamentos, regiões e territórios específicos. Ao analisar os tecidos
de indivíduos, a amostragem de tecido intraindividual pode revelar
mudanças de localização e dieta de forma semelhante à escala de semanas a
meses (por exemplo, Casar et al., 2018
), e quando os dados de múltiplos indivíduos são combinados, padrões
mais amplos podem ser discernidos. Práticas de amamentação e desmame de
indivíduos e populações ocorrem nesse nível (ver Waters-Rist, neste
volume), assim como estudos de história de vida de indivíduos quando
apenas amostras em massa são consideradas (Buikstra et al., 2004 ; Somerville & Braswell, 2016 ).
Em
uma escala macroscópica de décadas a séculos, os comportamentos humanos
podem ser investigados em termos de amplos processos sociais e
culturais, como normas culturais, diferenças de gênero, etnia e outros
aspectos macroscópicos das estruturas sociais humanas. Devido à sua
natureza abrangente, esses comportamentos humanos são melhor observados
em vastas regiões, territórios ou áreas culturais. Para abordar essas
questões por meio de isótopos, os pesquisadores podem analisar amostras
de tecido ósseo, particularmente osso e esmalte, de muitos indivíduos em
grandes áreas geográficas. Como o tecido ósseo se remodela ao longo da
vida, dependendo da idade do indivíduo e do elemento esquelético
analisado, uma amostra óssea pode representar a média das contribuições
isotópicas ao longo de um período de aproximadamente 10 a 20 anos
(Hedges et al., 2007
). Amostras de colágeno ou apatita óssea registram apenas as
características mais gerais da dieta ou do local de residência e,
portanto, são úteis principalmente para interpretações na escala de
mudanças populacionais ao longo do tempo. Em última análise, ao focar
claramente em escalas específicas de análise, padrões comportamentais,
tendências e variações em uma escala podem ser usados para compreender
os padrões em escalas mais amplas ou mais refinadas.
A
aplicação de estudos isotópicos na pesquisa arqueológica percorreu um
longo caminho desde sua introdução na década de 1970. O capítulo
introdutório deste volume, escrito por Margaret Schoeninger, discute o
desenvolvimento da área e como os estudos isotópicos se tornaram uma
ferramenta tão importante na pesquisa arqueológica. O epílogo, de Paul
Szpak, aborda o estado atual da área e suas possíveis direções futuras.
Os capítulos que se intercalam entre essas visões gerais abrangem uma
ampla gama de tópicos e destacam a diversidade atual do uso de métodos
isotópicos na pesquisa arqueológica.
Ao reunir uma coleção de
revisões e estudos de caso, este volume visa fornecer a estudantes,
arqueólogos e bioarqueólogos interessados em análises isotópicas uma
visão sobre como abordar questões relativas ao comportamento humano no
passado, compreendendo a escala e o alcance das ferramentas de pesquisa
disponíveis. Estudos isotópicos de materiais arqueológicos podem
contribuir para pesquisas sobre uma ampla gama de questões sociais e
comportamentais. Neste volume, apresentamos estudos de caso das três
áreas gerais de pesquisa para as quais eles têm sido mais produtivos:
(1) paleodieta e história de vida, (2) interações humano-animal e (3)
mobilidade e migração humana.
2.5.1 Paleodieta e História de Vida
Os
métodos isotópicos foram aplicados inicialmente a questões
arqueológicas relativas às práticas alimentares passadas tanto de
humanos (van der Merwe & Vogel, 1978 ; Vogel & van der Merwe, 1977 ) quanto de animais não humanos (Burleigh & Brothwell, 1978 ; Vogel, 1978
). Os valores de isótopos estáveis de carbono foram os primeiros a
serem empregados em estudos de paleodieta, mas o nitrogênio logo foi
adicionado ao conjunto de ferramentas (Schoeninger et al., 1983
). Combinadas, essas duas ferramentas isotópicas têm sido
revolucionárias em sua capacidade de fornecer dados quantitativos para o
estudo de dietas e histórias de vida passadas. Neste volume, as
contribuições de Waters-Rist, Bartelink et al., Salazar-Garcia et al. e
Rand demonstram como as ferramentas isotópicas podem ser integradas aos
estudos de paleodieta, fornecendo exemplos de meta-análises no espaço e
no tempo, testando os impactos das mudanças climáticas, utilizando
modelagem multi-isotópica e empregando novos tecidos biológicos.
Waters-Rist
apresenta uma revisão de dados isotópicos sobre alimentação infantil em
quase 100 populações ao longo dos últimos 11.000 anos e destaca, como
estudo de caso, dois grupos antigos da região do Lago Baikal, na
Sibéria. Waters-Rist enfatiza a plasticidade no processo de desmame,
conforme registrado por dados isotópicos, e argumenta que esses novos
métodos e interpretações refinadas permitirão uma melhor compreensão da
variação no comportamento humano no passado em relação a essa primeira e
crucial mudança alimentar na vida de um indivíduo. Bartelink e seus
colegas reconstroem paleodietas humanas para testar a hipótese de que
uma mudança climática na transição entre o Período Quente Medieval e a
Pequena Idade do Gelo impactou as estratégias de subsistência humana na
Ilha de Tutuila, Samoa Americana. Curiosamente, seus resultados
isotópicos demonstram a extensão da resiliência humana no uso de
recursos marinhos durante essa transição climática. Rand apresenta um
estudo de caso de uma abordagem multi-isotópica utilizando enxofre,
carbono, nitrogênio e oxigênio para ir além das reconstruções
paleodietéticas tradicionais na avaliação de quais fontes de recursos
vegetais e animais foram exploradas pelos antigos maias no sítio de
Caledônia. Salazar-Garcia e seus colegas examinam criticamente o
potencial do tártaro dentário como uma nova fonte de dados dietéticos
sobre indivíduos do passado. Seu capítulo descreve as vantagens de
incorporar o tártaro dentário como uma nova fonte de dados isotópicos
biológicos, revisa estudos anteriores sobre tártaro dentário e apresenta
um estudo de caso de uma população ancestral Puebloana Basketmaker II
do sudoeste americano para testar a eficácia do método.
2.5.2 Interações Humano-Animal
Os
comportamentos humanos do passado e as trajetórias históricas estão
frequentemente intimamente ligados aos dos animais que vivem dentro ou
perto de assentamentos humanos. Ao longo do tempo e do espaço, os
animais serviram como importantes fontes de alimento, trabalho, pele,
fibra, penas e companhia. Um dos primeiros estudos isotópicos em
arqueologia demonstrou que cães peruanos antigos consumiam altos níveis
de milho, semelhantes aos de seus donos humanos (Burleigh &
Brothwell, 1978
). A maioria dos outros estudos iniciais, no entanto, concentrou-se nas
paleodietas humanas até o final da década de 1980, quando os
pesquisadores começaram a se concentrar mais em amostras de fauna e em
questões de interação humano-animal sob a perspectiva do manejo e
fornecimento de alimentos para animais (por exemplo, DeNiro, 1988
). Neste volume, as contribuições de Kinaston, Somerville, Guiry e
colegas, e Swift fornecem revisões e estudos de caso abrangendo
múltiplos sistemas isotópicos e espécies animais para explorar como os
arqueólogos podem interpretar a relação entre os humanos e os animais
que eles manejaram, consumiram e moldaram por meio de mudanças
antropogênicas nas paisagens ecológicas.
As contribuições de
Kinaston e Somerville fornecem revisões abrangentes do uso de dados
isotópicos para melhor compreender as relações entre humanos e animais
ao longo do tempo. Já o capítulo de Kinaston tem um escopo global e
inclui múltiplos sistemas isotópicos (δ 13 Cd 15 N, d 34 S, d 18 O, e 87 Sr./ 86 Sr.),
o capítulo de Somerville concentra-se exclusivamente nas relações entre
humanos e animais nas Américas e discute como um único isótopo, δ 13 O
δ¹³C tem sido usado para elucidar práticas de manejo e criação nas
Américas durante o período pré-colonial. As contribuições de Guiry e
colegas e de Swift apresentam estudos de caso específicos que destacam a
utilidade dos estudos de isótopos estáveis em restos faunísticos para
elucidar questões mais amplas sobre o impacto humano em espécies
animais. Guiry e colegas analisaram restos de porcos ( Sus domesticus ) de uma vila de pescadores do século XIX na Islândia para determinar o δ¹³C. 13 C e δ 15 Os
valores de N, numa tentativa de determinar se os porcos foram criados
localmente ou importados, como sugerido em fontes históricas, revelam
evidências de produção local. Os resultados são relevantes não apenas
para a história local da relação entre humanos e porcos na Islândia, mas
também para questões mais amplas, como a natureza das redes de comércio
transatlântico. Swift utiliza a análise de isótopos estáveis de
restos de ratos-do-pacífico ( Rattus exulans ) da ilha
polinésia de Mangareva para explorar as mudanças antropogênicas no
ecossistema da ilha. Os ratos chegaram às ilhas juntamente com os
humanos e, com o tempo, proliferaram rapidamente, provavelmente
contribuindo para o declínio da biodiversidade local. A análise de
isótopos estáveis de δ 13 C e δ 15 Os valores de
N obtidos a partir de ossos de ratos lançam luz sobre como a paisagem
da ilha mudou ao longo do tempo e de que forma o rato do Pacífico pode
ser usado como um indicador do impacto antropogênico dentro de um
ecossistema.
2.5.3 Mobilidade Humana e Migração
A
mobilidade e a migração têm sido fatores importantes nas sociedades
humanas há muito tempo. O movimento de pessoas pela paisagem pode
ocorrer como parte das mudanças sazonais nas estratégias de subsistência
das comunidades, por indivíduos que mudam de residência por motivos de
casamento ou oportunidades econômicas, ou por populações inteiras
deslocadas por guerras, fome ou desastres naturais (Anthony, 1997 ; Arnauld et al., 2021 ; Tilly, 1978
). Embora as primeiras aplicações do estrôncio na arqueologia tenham se
concentrado no uso das razões Sr/Ca para inferir a paleodieta
(Schoeninger, 1979 ), foi Ericson ( 1985 ) quem reconheceu que a 87 Sr./ 86 A
proporção de Sr em ossos adultos em comparação com os valores dentários
em amostras arqueológicas pode identificar indivíduos migrantes dentro
de uma população. As décadas de 1990 e início de 2000 produziram
diversos artigos sobre uma variedade de animais não humanos,
estabelecendo características locais. 87 Sr./ 86 Os
valores de Sr em todo o mundo foram ampliados à medida que o método se
expandiu para o conjunto de ferramentas arqueológicas (ver revisão da
literatura em Bentley, 2006
). Dois capítulos deste volume utilizam métodos isotópicos para
explorar a movimentação de povos antigos pela paisagem, e ambos
apresentam estudos de caso com foco em sociedades da América do Sul
pré-colonial.
Knudson e Torres usam δ 18 O e 87 Sr./ 86 Lucas
e seus colegas utilizam dados de estrôncio extraídos de restos humanos
dos oásis de San Pedro de Atacama, no norte do Chile, para argumentar
que estudos detalhados e de longo prazo podem fornecer perspectivas
únicas sobre a migração. Eles discutem seus dados em relação às mudanças
políticas e ambientais em larga escala da época, situando as evidências
de mobilidade humana como uma possível resposta adaptativa a mudanças
contextuais mais amplas. 87 Sr./ 86 Dados de Sr de
ossos e dentes humanos foram utilizados para explorar a natureza da
interação cultural no Vale de Cochabamba, na região central da Bolívia.
Como o público-alvo deste volume são estudantes e arqueólogos, o pequeno
tamanho da amostra ( n = 6) deste estudo destaca o potencial
de um planejamento de pesquisa direcionado para ilustrar o comportamento
humano no passado por meio de estudos isotópicos. Os resultados revelam
a presença de indivíduos nascidos no exterior na população local,
indicando que essa comunidade mantinha laços significativos com centros
populacionais distantes.
2.6 Conclusão
Hoje,
a análise isotópica é um método comumente aplicado no conjunto de
ferramentas de arqueólogos e bioarqueólogos, graças aos avanços na
automação da tecnologia, que tornam sua aplicação mais rápida e fácil do
que nunca. Como discutido por Szpak no epílogo deste volume, a formação
de uma nova geração de especialistas em isótopos exige que eles sejam
geradores de dados altamente competentes. Mas, assim como Szpak, também
reconhecemos que nem todo usuário de dados isotópicos precisa ser o
gerador primário ou especialista; em vez disso, pode ser um usuário
final experiente dos dados. O objetivo deste volume é ajudar estudantes e
arqueólogos a aprimorar e expandir suas possibilidades de pesquisa por
meio da teoria, de novas aplicações da ferramenta e da compreensão das
diferentes escalas de análise mais apropriadas para questões específicas
sobre o comportamento humano no passado. Os estudos de caso a seguir
destacam a eficácia da pesquisa arqueológica orientada por perguntas,
que coloca os estudos isotópicos como o método central de análise. Como
enfatizado por Schoeninger no capítulo introdutório deste volume, o
estudo do comportamento humano no passado é um campo impulsionado tanto
por ideias quanto por ferramentas. Ficamos satisfeitos que esta
coletânea de artigos destaque a diversidade tanto das ideias de pesquisa
quanto das ferramentas isotópicas atualmente utilizadas para desvendar
as complexidades do passado da humanidade.
Referências
Agrawal, M. (2003). Respostas das plantas ao enxofre atmosférico. Em YP Abrol & A. Ahmad (Eds.), Enxofre em plantas (pp. 279–293). Springer. https://doi.org/10.1007/978-94-017-0289-8_15
Ambrose, SH (1990). Preparação e caracterização de colágeno ósseo e dentário para análise isotópica. Journal of Archaeological Science, 17 , 431–451. https://doi.org/10.1016/0305-4403(90)90007-R
Ambrose,
SH, & Norr, L. (1993). Composição isotópica de proteína e energia
dietética versus colágeno e apatita óssea: experimentos de crescimento
com dieta purificada. Em JB Lambert & G. Grupe (Eds.), Arqueologia molecular do osso humano pré-histórico (pp. 1–37). Springer.
Amundson,
R., Austin, AT, Schuur, EAG, Yoo, K., Matzek, V., Kendall, C.,
Uebersax, A., Brenner, D., & Baisden, WT (2003). Padrões globais da
composição isotópica do solo e do nitrogênio das plantas. Ciclos Biogeoquímicos Globais, 17 , 1031. https://doi.org/10.1029/2002gb001903
Anthony, DW (1997). Migração pré-histórica como processo social. Em J. Chapman & H. Hamerow (Eds.), Migrações e invasões na explicação arqueológica . Archaeopress, BAR S664, Oxford.