Genoma do abacaxi bracteatus e domesticação de culturas propagadas por clones
Nature Genetics, volume 51 , páginas 1549-1558 ( 2019 )
Abstrato
A hipótese de que a domesticação de culturas propagadas clonalmente,
como o abacaxi da América do Sul, era uma "operação de uma etapa". Sequenciamos o genoma de Ananas comosus var. bracteatus CB5 e reuniu 513 Mb em 25 cromossomos com 29.412 genes.
A comparação dos genomas de CB5, F153 e MD2 elucidou a base genômica da
produção de fibras, formação de cores, acúmulo de açúcar e maturação
dos frutos. Também ressequenciamos 89 genomas de ananás .
Os cultivares 'Smooth Cayenne' e 'Queen' exibiram mistura antiga e
recente, enquanto o 'Singapore Spanish' apoiou uma operação de
domesticação em uma etapa.
Identificamos 25 varreduras seletivas, incluindo uma varredura forte
contendo um par de inibidores de bromelina duplicados em conjunto. Quatro genes candidatos à auto-incompatibilidade foram ligados no F153, mas não eram funcionais no CB5 auto-compatível.
Nossas descobertas apóiam a coexistência de recombinação sexual e uma
operação em uma etapa na domesticação de culturas propagadas por clones. Este trabalho orienta a exploração de trajetórias de domesticação sexual e assexuada em outras culturas propagadas por clones.
a Principal
A maioria das culturas de grãos, vegetais e plantas ornamentais é
produzida sexualmente através da propagação de sementes, enquanto a
maioria das árvores frutíferas, tubérculos e algumas plantas ornamentais
são clonais propagadas através de enxertos, cultura de tecidos,
divisões ou estacas. As espécies que se reproduzem sexualmente passam por centenas a milhares de gerações de recombinação durante a domesticação; essa seleção recorrente deixa assinaturas altamente tratáveis no genoma.
Por outro lado, a domesticação de culturas propagadas clonicamente
depende da reprodução vegetativa e sexual, agindo esporadicamente nos
clones de vida longa. Pode até ser uma operação de uma etapa, em que a seleção é concluída depois que um clone é selecionado 1 .
Portanto, as culturas clonais podem ter passado de zero a alguns ciclos
de recombinação e seleção pós-domesticação, em nítido contraste com as
culturas anuais reproduzidas sexualmente.
O abacaxi ( Ananas comosus (L.) Merr.) É uma cultura de frutas originada e domesticada na América do Sul. Segundo Bertoni 2 , o nome do gênero Ananas significa 'excelente fruto' na língua guarani do Paraguai.
O abacaxi foi domesticado> 6.000 anos atrás, com restos arqueobotânicos datados de 3.500 anos atrás na América do Sul e distribuídos para a Mesoamérica> 2.500 anos atrás 3 , 4 , 5 . O abacaxi é propagado clonalmente usando a coroa de frutas folhosas, enxertos ou ventosas.
O abacaxi vermelho ( Ananas comosus var. Bracteatus ) era cultivado anteriormente para fibra, suco de frutas e como uma cobertura viva, e agora é um ornamental pantropical 6 , 7 . A planta bracteatus é notável por seus frutos de cor rosa-avermelhada brilhantes.
O nome ' bracteatus ' refere-se às suas longas brácteas. A planta é vigorosa com folhas longas, espinhos grossos e ventosas abundantes. As fibras vegetais têm sido usadas em inúmeras aplicações benéficas para a agricultura e o meio ambiente, em parte devido à sua natureza biodegradável e falta de carcinogenicidade. A fibra da folha de abacaxi (PALF) contém 70 a 82% de celulose, 5 a 12% de lignina e 1,1% de cinza 8 . O PALF é uma importante fonte de fibra natural e tem sido utilizado na produção de fibras de carvão ativado 9 , materiais de embalagem 10 , andaimes celulares 11 e aparelhos 12 .
O abacaxi está em Bromeliaceae, que inclui> 3.000 espécies agrupadas em> 50 gêneros 13 , 14 . As bromélias desafiam os conceitos clássicos de espécies por causa de suas barreiras pré e pós-zigóticas notoriamente vazantes 15 , 16 , 17 .
O ananás é único na família por seus frutos sinárpicos. A variedade bracteatus é um cultivigen, cultivado anteriormente para fibras no sudeste do Brasil, Paraguai e norte da Argentina. Aqui, geramos um segundo genoma de referência de Ananás a partir da adesão de bracteatus CB5 e refazemos numerosas cultivares de abacaxi e espécies selvagens de Ananás para explorar a diversidade e a história de domesticação do abacaxi, padrões de propagação clonal e assinaturas de seleção humana.
O abacaxi ( Ananas comosus (L.) Merr.) É uma cultura de frutas originada e domesticada na América do Sul. Segundo Bertoni 2 , o nome do gênero Ananas significa 'excelente fruto' na língua guarani do Paraguai.
O abacaxi foi domesticado> 6.000 anos atrás, com restos arqueobotânicos datados de 3.500 anos atrás na América do Sul e distribuídos para a Mesoamérica> 2.500 anos atrás 3 , 4 , 5 . O abacaxi é propagado clonalmente usando a coroa de frutas folhosas, enxertos ou ventosas.
O abacaxi vermelho ( Ananas comosus var. Bracteatus ) era cultivado anteriormente para fibra, suco de frutas e como uma cobertura viva, e agora é um ornamental pantropical 6 , 7 . A planta bracteatus é notável por seus frutos de cor rosa-avermelhada brilhantes.
O nome ' bracteatus ' refere-se às suas longas brácteas. A planta é vigorosa com folhas longas, espinhos grossos e ventosas abundantes. As fibras vegetais têm sido usadas em inúmeras aplicações benéficas para a agricultura e o meio ambiente, em parte devido à sua natureza biodegradável e falta de carcinogenicidade. A fibra da folha de abacaxi (PALF) contém 70 a 82% de celulose, 5 a 12% de lignina e 1,1% de cinza 8 . O PALF é uma importante fonte de fibra natural e tem sido utilizado na produção de fibras de carvão ativado 9 , materiais de embalagem 10 , andaimes celulares 11 e aparelhos 12 .
O abacaxi está em Bromeliaceae, que inclui> 3.000 espécies agrupadas em> 50 gêneros 13 , 14 . As bromélias desafiam os conceitos clássicos de espécies por causa de suas barreiras pré e pós-zigóticas notoriamente vazantes 15 , 16 , 17 .
O ananás é único na família por seus frutos sinárpicos. A variedade bracteatus é um cultivigen, cultivado anteriormente para fibras no sudeste do Brasil, Paraguai e norte da Argentina. Aqui, geramos um segundo genoma de referência de Ananás a partir da adesão de bracteatus CB5 e refazemos numerosas cultivares de abacaxi e espécies selvagens de Ananás para explorar a diversidade e a história de domesticação do abacaxi, padrões de propagação clonal e assinaturas de seleção humana.
Resultados
Montagem do genoma e anotação da adesão de abacaxi bracteatus CB5
O tamanho do genoma do CB5 foi estimado em ~ 591 Mb por citometria de fluxo. Geramos 26,9 Gb de leituras da plataforma PacBio RSII e ~ 100 × Illumina de leituras curtas. A montagem inicial usando CANU produziu 809,6 Mb de sequência montada. Para eliminar sequências homozigotas redundantes, desenvolvemos um novo algoritmo, Pseudohaplóide, que identifica e filtra contigs heterozigotos com base no alinhamento de todo o genoma. O conjunto resultante foi de 513 Mb, com um N50 contig de 427 kb com 92,6% de completude e sequências duplicadas muito reduzidas (Tabelas Suplementares 1 e 2 ). O alinhamento dos transcritos montados no sequenciamento de RNA (RNA-seq) ao genoma revelou 99,92% de identidade de sequência (Tabela Suplementar 3 ). Além disso, 98,47% das leituras de Illumina estavam alinhadas ao genoma, cobrindo 99,51% do genoma (Tabela Suplementar 4 ).Os contigs foram corrigidos e estruturados por captura de conformação de cromatina de alto rendimento (Hi-C) em 25 pseudo-cromossomos que ancoravam 456 Mb (88,8%) do genoma (Fig. 1 , Fig. 1 e Tabela 5 ). No geral, 29.412 modelos de genes codificadores de proteínas putativos foram anotados (Tabela Suplementar 6 ). Identificamos 383,2 Mb de seqüências repetitivas, responsáveis por 74,7% do genoma montado (Figura Suplementar 2 e Tabela Suplementar 7 ). As distâncias de Kimura indicaram uma explosão de longa atividade de retrotransposão terminal repetida (LTR-RT) ~ 1,8 milhão de anos atrás.
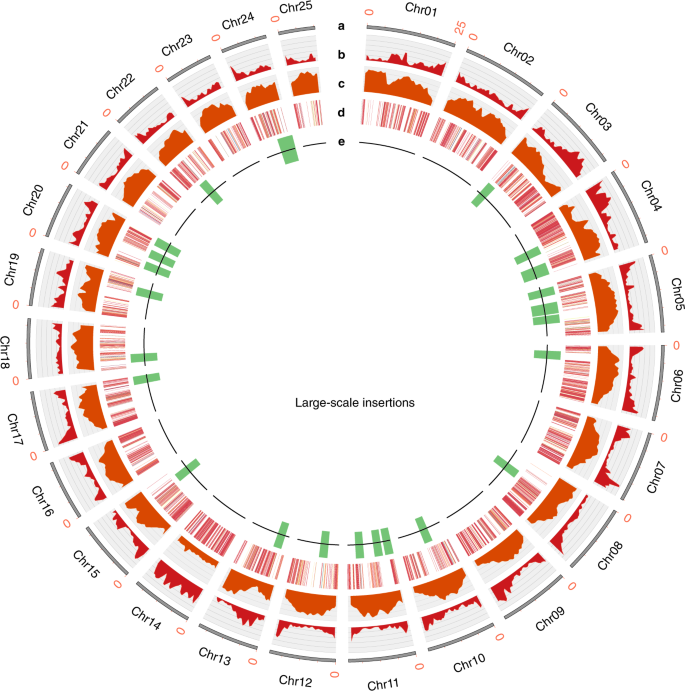
a - e , Os anéis indicam (do mais externo ao mais interno) 25 cromossomos ( a ), densidade gênica ( b ), abundância de elementos transponíveis ( c ), variação do número de cópias gênicas ( d ) e inserções em larga escala em comparação com o genoma F153 ( e )
Montagem aprimorada do genoma do abacaxi F153
O maior conteúdo de LTR-RT cigana está concentrado próximo aos centrômeros nas angiospermas 18 . A distribuição dos elementos ciganos foi plotada ao longo das 25 pseudomoléculas no genoma F153 (referido como F153 v.6) 19 . Dois picos foram observados no grupo de ligação (LG) 01 (Fig. 3 Complementar), uma pseudomolécula quimérica correspondente a dois cromossomos no CB5 (Fig. Suplementar 4 ). Havia um pico cigano em uma extremidade do LG24, enquanto não havia região rica em ciganos no LG25, que se alinhava a um cromossomo no CB5. Os LGs desmontados foram corrigidos no conjunto do genoma F153 aprimorado (referido como F153 v.7), no qual LG01 foi separado em AccChr1 e AccChr24, enquanto LG24 e LG25 foram vinculados no AccChr25.Base genômica da produção de fibras no abacaxi CB5
Tanto o F153 como o CB5 têm oito genes CesA (Tabela Suplementar 8 ), agrupados nos necessários para a biossíntese da parede celular primária ( CesA1, 3, 6 e 9 ) e secundária ( CesA4, 7, 8 e 11 ). Os genomas CB5 e F153 compartilham os mesmos genes, mas não possuem ortólogos dos genes CesA2 , 5 e 10 em Arabidopsis (Fig. 5 complementar). Em F153 e CB5, os genes para a biossíntese da parede celular primária foram todos altamente expressos em folhas, flores e frutos, exceto no CesA9 . Curiosamente, os genes CesA4 , 7 e 8 , que estão envolvidos na síntese da parede celular secundária, foram altamente expressos nas folhas de F153, enquanto seus níveis de expressão eram baixos no CB5 (Figura 6 ).A lignina é o segundo componente principal do PALF. O conjunto completo de genes biossintéticos de lignina de abacaxi foi identificado por alinhamento de sequência aos genes conhecidos da via de síntese de Arabidopsis , arroz e álamo 20 , 21 (Conjunto de dados suplementar 1 ). CB5 e F153 tinham 24 e 21 genes candidatos para a biossíntese de lignina, respectivamente. Três genes PAL no CB5 apresentaram maior expressão nas folhas do que no F153 (Figura 7 ). Tanto o COMT1 quanto o CCOMT1 apresentaram maior expressão no CB5 do que no F153 (Figura 8 ).
Genes biossintéticos da antocianina
A variedade bracteatus é frequentemente cultivada como planta ornamental, em parte devido à cor vermelha de seus frutos. A biossíntese da antocianina compartilha a via dos fenilpropanóides com a biossíntese da lignina em seus primeiros passos. Os genes biossintéticos da antocianina foram identificados em F153 e CB5 (Tabela Suplementar 9 ). O tamanho das famílias de genes CB5 que codificam genes biossintéticos de antocianina foi maior do que em F153 (22 versus 17). Genes biossintéticos iniciais na via como CHS , CHI , F3H e F3 ′ H foram expandidos no CB5. Tanto o F153 como o CB5 não possuíam ortólogos FLS e ANS , indicando a existência de seus genes de isozima, que podem assumir suas funções.Genes do metabolismo do açúcar
A doçura é uma característica importante da qualidade da fruta. Nos abacaxis, a sacarose é o principal açúcar, seguida pela glicose e frutose 22 . Várias enzimas participam de sua biossíntese, transporte e metabolismo, sem diferença no número de genes entre CB5 e abacaxi, incluindo sacarose-fosfato sintase, sacarose-fosfato fosfatase, sacarose-sintase, invertases, sacarose transportadores (SUTs), açúcares que eventualmente transportadores não exportados (SWEETs) e transportadores de monossacarídeos 23 , 24 , 25 , 26 (Tabela Suplementar 10 ). No CB5, os SUTs foram expressos constantemente em um nível baixo durante a maturação dos frutos (Tabela Suplementar 11 ), enquanto dois dos genes SUT ( AccSUT1 e AccSUT3 ) foram altamente expressos em frutos maduros no MD2 (Tabela Suplementar 12 ). Mais genes SWEET foram expressos no estágio de desenvolvimento tardio dos frutos no MD2 do que no CB5 (Tabelas Suplementares 13 e 14 ). Mais interessante, o AccSWEET13 estava localizado na região do genoma F153, onde foi detectada uma varredura seletiva (veja abaixo). Esses resultados explicam parcialmente porque o MD2 acumula mais açúcar em seus frutos que o CB5.Bromelinas
Foram identificadas 61 e 47 bromelinas do tipo cisteína proteinase (CP) em F153 e CB5, respectivamente. Enquanto isso, identificamos 28 CPs em Amborella , 36 em Arabidopsis , 34 em mamão, 25 em uva, 50 em álamo, 47 em sorgo e 50 em arroz (Tabela Suplementar 15 ). Esses PCs são divididos em nove subfamílias (Figura 9 ). A subfamília VI tinha mais membros, enquanto as subfamílias V, VIII e IX tinham menos membros, com não mais de três membros em cada espécie. Observou-se expansão na subfamília VI em todas as espécies selecionadas, principalmente F153. As bromelinas de abacaxi pertencem a esta subfamília e a expansão pode resultar em uma alta produção de bromelinas. A maioria dos PCs mostrou padrões de expressão constantes durante o amadurecimento dos frutos (Tabelas Suplementares 16 e 17 ). Alguns genes, como AccCEP3 e AccPAP25, apresentaram padrões de expressão dinâmica em alto nível durante o estágio maduro do amadurecimento dos frutos (Tabela Suplementar 16 ). Na subfamília VI de CB5, apenas dois genes apresentaram expressão nos tecidos estudados. Verificou- se que o AcbPAP10 era expresso em flores, frutos e folhas, enquanto o AcbPAP17 era expresso apenas em flores. Genes mais expressos na subfamília VI foram detectados em F153. AccPAP3 e AccPAP4 exibiram uma expressão muito alta nos estágios finais do desenvolvimento dos frutos, talvez contribuindo para o amadurecimento dos frutos.Padrões de variação em todo o genoma do abacaxi
Foram selecionados 89 acessos de Ananás para o sequenciamento de todo o genoma, incluindo 67 acessos de A. comosus var. comosus , nove acessos de var. bracteatus , dois acessos de var. erectifolius , nove acessos da variedade selvagem. microstachys e dois acessos de Pitcairnia gracilis e P. punicea como subgrupos (Fig. 2 e Tabela Suplementar 18 ). O var. As amostras de comosus incluem representantes das três cultivares históricas 'Queen', 'Smooth Cayenne' e 'Singapore Spanish', associadas à difusão pantropical do abacaxi nos tempos históricos. Outra var. Importante As cultivares de comosus analisadas incluem 'Pérola' e várias cultivares do noroeste da América do Sul e América Central, bem como linhagens reprodutivas e cultivares de origem desconhecida. Também incluímos clones cultivados de A. comosous var. bracteatus e o progenitor selvagem proposto do abacaxi A. comosus var. microstática 27 (Tabela Suplementar 19 ).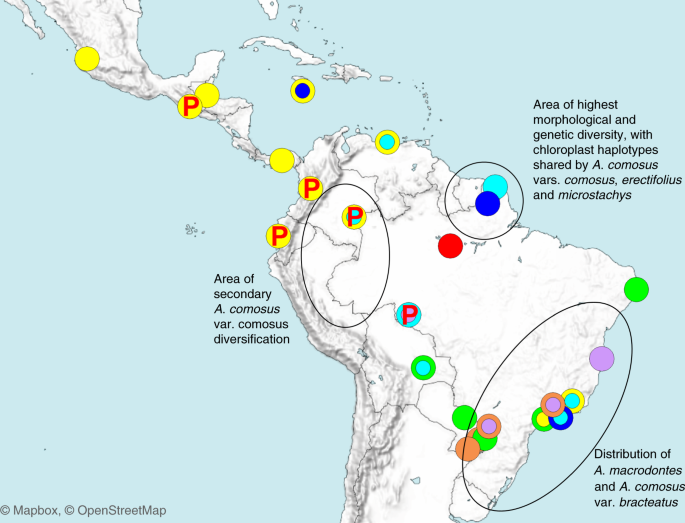
Os acessos são representados pelo mesmo código de cores da análise de mistura (Fig. 3 ). Os acessos com menos de 10% de mistura são representados pela cor do seu cluster.
Para acessos misturados, a cor que vem em primeiro lugar em importância
no diagrama de mistura é indicada no círculo concêntrico mais amplo e a
segunda em importância no círculo concêntrico menor: azul claro,
cluster Cayenne liso de var. comosus ; azul escuro, grupo da rainha; violeta, cluster espanhol de Cingapura; amarelo, quarto agrupamento (cultivares relacionadas a Mordilona); vermelho, var. cluster erectifólio ; laranja, var. cluster bracteatus ; verde, var. cluster microstachys . Cultivares portadores do gene supressor de coluna 'tubulação' são identificados por uma letra P vermelha.
Os espectros de frequência de sites não-sinônimo e sinônimo foram examinados para as cultivares Smooth Cayenne, Queen e Singapore Spanish (Fig. 11 complementar). O Cayenne suave possui um excesso de variantes não sinônimas de baixa frequência em comparação com variantes sinônimas, indicando a seleção purificadora. Para os espanhóis Queen e Cingapura, havia um excesso incomum de variantes em uma frequência intermediária para sites não-sinônimo e sinônimo. Provavelmente porque os espanhóis Queen e Cingapura tinham uma abundância maior de genótipos heterozigotos por posição de SNP (Fig. 12 complementar).
Origem, estrutura populacional e ancestralidade genômica do abacaxi
Utilizamos um subconjunto de 665.162 SNPs de qualidade filtrada para explorar as relações entre os genomas de taxa e cultivares divergentes de Ananas . Árvores e redes filogenéticas estimadas com RAxML 28 e SplitsTree 29 separaram acessos das variedades microstachys , bracteatus , erectifolius e comosus e acessos de cultivares importantes dentro deste último. Sete cultivares mal rotuladas foram corrigidas e seis cultivares foram atribuídas a cultivares corretas que não podiam ser classificadas anteriormente (Tabela Suplementar 18 ). Para Cingapura Espanhola e Selangor Green, confirmamos e completamos a história de sua difusão do leste do Brasil para a Ásia. Da mesma forma, os dois var. acessos de erectifolius foram obtidos da mesma coleção original por propagação vegetativa (Fig. 3a , Tabela Suplementar 18 e Figs. 13 e 14 Suplementares).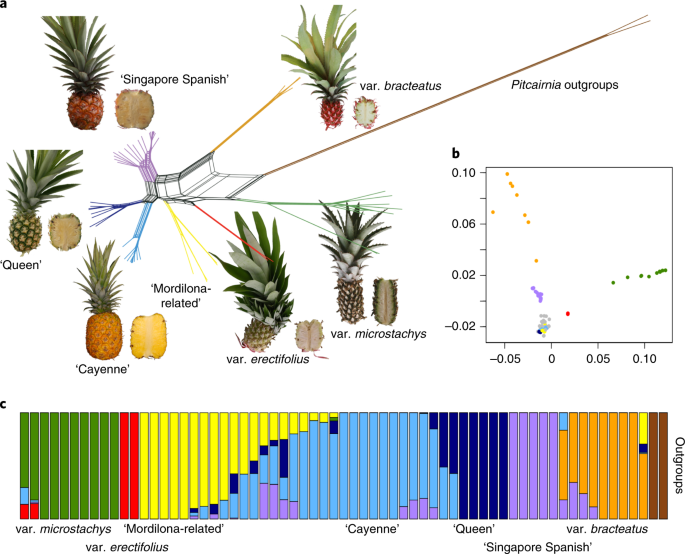
a , rede SplitsTree para acessos a Ananas, excluindo amostras misturadas. Verde, microestaca de variedade; vermelho, variedade erectifolius ; laranja, variedade bracteatus ; amarelo, variedade comosus / cultivares relacionadas a Mordilona Cambray / Monte Lirio; roxo, variedade comosus / cultivar Cingapura Espanhol; azul claro, variedade comosus / cultivar Smooth Cayenne; azul escuro, variedade comosus / cultivar Queen; marrom, gênero Pitcairnia outgroups. Uma rede de amostras misturadas é mostrada na Fig. Suplementar 14 . b , gráficos MDS dos acessos de Ananás estudados, com eixos horizontal e vertical explicando 33,0% e 20,6% da variância, respectivamente. O código de cores segue que em um genótipo de A. comosus misturado e indicado em cinza. c , a ascendência resulta de ADMIXTURE sob o modelo K = 8, suportado por um exame de erros de validação cruzada (Fig. 15 complementar).
Os padrões de diversidade e relacionamento foram confirmados por MDS dos dados genômicos (Fig. 3b e Fig. 19 ). Os comprimentos dos ramos das árvores envolvendo as variedades microstachys , bracteatus , erectifolius e comosus foram comparados àqueles entre as principais cultivares de comosus (Fig. 3a ). A divergência genômica absoluta ( D xy ; Fig. 16 suplementar) foi significativamente maior entre pares de variedades, em comparação com as principais cultivares de comosus ( P <0 span=""> D xy entre as variedades de Ananás foi em média 0,0059 (mediana 0,0046, sem 0,0007), o que está dentro do intervalo de expectativas para espécies derivadas recentemente 31 , 32 .
A ancestralidade genética local de acessos híbridos estimada com uma abordagem do Modelo Markov Oculto revelou uma grande diversidade de padrões, incluindo híbridos com grandes segmentos de ancestrais provenientes de diferentes cultivares modernas de comosus e híbridos com pequenos segmentos de ancestrais de diferentes cultivares e táxons (Fig. 4 ). A presença de segmentos grandes e pequenos em híbridos indicou que a mistura afetou a evolução da variedade comosus em longas escalas de tempo. Nossos modelos mais prováveis de ascendência local eram consistentes com uma média de 37 gerações desde o início da mistura (variação de 21 a 55) entre as 22 var. híbridos comosus detectados em nosso estudo. Para as microestacas da variedade selvagem, as estimativas individuais variam de 107 a 612 gerações, respectivamente. Esses números provavelmente se traduziram em vários milhares de anos como plantas perenes, principalmente de propagação assexuada.
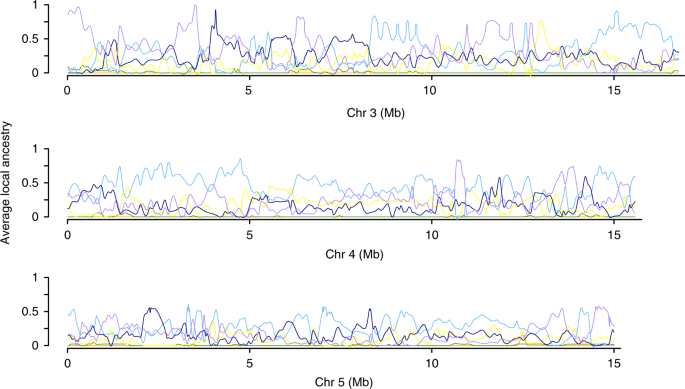
Regiões genômicas com proporções de ancestralidade incomumente altas de
variedades particulares de abacaxi são visíveis ao longo dos
cromossomos. Os cromossomos restantes mostram resultados semelhantes. Para etiquetas de código de cores, consulte a Fig. 3 . Todos os valores de ascendência somam 1 para cada janela genômica.
Assinaturas genômicas de seleção mitótica e propagação clonal
A mutação somática é a principal força motriz que molda a domesticação e diversificação de plantas propagadas clonicamente 33 . Uma fonte de mutação somática é o movimento de elementos transponíveis (TEs). Pesquisamos a presença / ausência de variação de pequenos DNA EEs em 89 acessos reequilibrados. As ETs de DNA eram altamente abundantes, atingindo números de cópias de até dezenas de milhares 34 e se inserem predominantemente em regiões ricas em genes ou nas proximidades 35 . O software MITE-Hunter previu 4.614 junções TE consistindo em 2.286 Mutator , 1.156 hAT, 1.018 PIF / Harbinger, 128 CACTA e 26 elementos desconhecidos. Os locais de junção exclusivos criados pelas inserções de TE foram usados como referência para o mapeamento de leitura para avaliar a presença / ausência de variação em relação à referência F153. No total, 98.476 junções TE foram identificadas na montagem de abacaxi de referência: 46.613 Mutator , 23.634 PIF / Harbinger, 18.831 hAT, 4.091 CACTA, 254 incógnitas e 12 junções formadas por duas superfamílias TE diferentes. Comparado ao genoma de referência F153, cada acesso exibia um grande número de junções TE únicas, que variavam de 97% de identidade com F153 em Ac50 a 28% de identidade com F153 em Ac46c (Tabela Suplementar 22 ). A alta variabilidade dos locais de inserção da TE no abacaxi pode ser um fator determinante para novas características por meio de mutação somática durante a domesticação.Prevê-se que o processo de recombinação mitótica leve à homozigose terminal ao longo do tempo em tecidos ou organismos propagados exclusivamente por meios somáticos. Essa geração aleatória de homozigose em tecidos inicialmente heterozigotos 36 poderia fornecer variação genética selecionável ao descobrir alelos recessivos. Por isso, investigamos essa questão encontrando primeiro todos os genes de cópia única (SC) no genoma do abacaxi. Os genes SC foram escolhidos para que a identificação de heterozigosidade versus homozigose para qualquer localização cromossômica específica pudesse ser determinada sem a confusão gerada pelos paralogs. Os 10.439 genes SC finais foram distribuídos aleatoriamente pelo genoma (Fig. 20 complementar).
As execuções terminais de homozigose nas extremidades dos LGs eram frequentemente detectadas, especialmente em espanhol de Cingapura, incluindo LG01, 03, 04, 08, 11, 14, 15, 20, 22 e 24 (Fig. 5 e Fig. 21 ). Parte dessa homozigose abrangeu toda a região, desde o local da recombinação mitótica até o final do cromossomo, como esperado 37 . A presença de tais execuções terminais de homozigose indicou uma ocorrência precoce (e possível seleção e fixação) de mutações mitóticas associadas no processo de domesticação. Em Smooth Cayenne e Queen, a homozigose terminal curta foi detectada esporadicamente nos LG03 e 23 e era provável que fosse um produto de reprodução clonal e sexual mista. Notavelmente, o nível geral de heterozigosidade (e a falta de quase um pequeno número de regiões homozigotas) nos parentes selvagens de abacaxi indicavam que essas populações eram cruzadores prodigiosos.
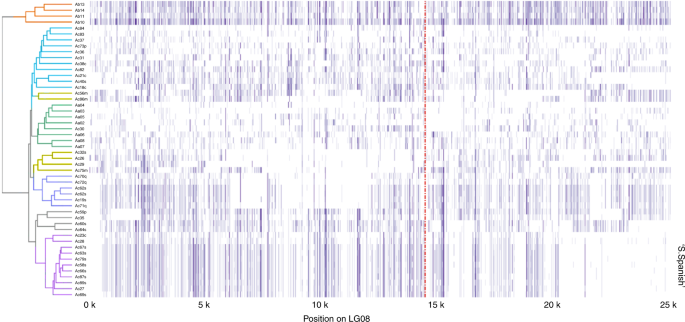
Níveis
de heterozigosidade foram plotados para cada 100 pb através de genes
concatenados linearmente e através de 50 acessos com baixos níveis de
mistura. Um mapa de calor
da heterozigosidade é plotado em que o branco não indica
heterozigosidade e o roxo escuro indica alta heterozigosidade. A
linha vermelha pontilhada vertical indica a região prevista do
centrômero, conforme determinado pela abundância de retrotransposão
Gypsy LTR. Os dendrogramas à esquerda indicam paisagens heterozigoticas agrupadas entre as variedades.
Varreduras seletivas e seleção de formas de origem sexual durante a domesticação de abacaxi
As regiões genômicas de seleção durante a domesticação do abacaxi foram identificadas com base em reduções drásticas na diversidade de nucleotídeos ( π ) nos acessos cultivados, em comparação com as linhas selvagens ( π c / π w ) nas janelas deslizantes do genoma. A diversidade dentro de microestacas foi utilizada para estimar π w. As cultivares com evidência de mistura foram omitidas nas varreduras de seleção e π cfoi calculado dentro e através de cada uma das quatro cultivares. As regiões varridas por candidatos foram ainda mais estreitadas usando uma abordagem baseada no teste de razão de verossimilhança composta entre populações (XP-CLR) para modelar as diferenças no espectro de frequências de alelos entre acessos cultivados e selvagens. Essa abordagem identificou 25 supostas varreduras de domesticação no genoma do abacaxi com tamanhos variando de 150 kb a 1,2 Mb (Tabela Suplementar 23 ). As regiões varridas cobriram coletivamente 11,9 Mb (~ 3,1% do genoma), o que foi substancialmente menor do que os padrões observados em culturas sexualmente propagadas, como tomate (186 varreduras de domesticação totalizando 64,5 Mb, ref. 38 ) e soja (121 varreduras totalizando 53 Mb, ref. . 39) O abacaxi também teve menos varreduras seletivas putativas do que outras culturas propagadas por clones, como a mandioca, que contém assinaturas de 224 varreduras 40 .As regiões varridas no abacaxi englobaram 392 genes com enriquecimento nas vias de resposta ao estresse (FDR = 2,1 × 10-3 ), mas nenhum enriquecimento óbvio em genes previamente caracterizados em outras espécies como responsáveis por características relacionadas à domesticação. Para restringir esta lista de genes candidatos à domesticação, pesquisamos alterações na expressão gênica em uma série de alta resolução de frutos de abacaxi em desenvolvimento. A varredura mais forte foi uma região de 225 kb no início do LG03, com uma redução de 400 vezes na diversidade entre acessos cultivados em comparação com a var selvagem. microstáticas (Fig. 6a ). Este varrimento foi no topo de 5% com base no XP-CLR que indicado baixo F ST e altamente negativa de Tajima D (Fig. 6b, c) Embora a varredura no LG03 se sobreponha a um longo período de homozigose terminal (Fig. 21.3 Suplementar ), foi muito mais estreita que o ciclo de homozigose (Fig. 5 ). A varredura putativa contém nove genes, incluindo um par de inibidores de bromelina duplicados em conjunto ( AccBI1 e AccBI2 ) com padrões de expressão específicos para frutas (Fig. 6d ). As bromelinas em coordenação com os inibidores da bromelina devem desempenhar um papel importante no amadurecimento dos frutos de abacaxi 41 , 42 . O inibidor da bromelina é inativado pós-traducionalmente durante o amadurecimento do fruto, levando a um aumento significativo na atividade da bromelina, melhorando a proteólise, o amolecimento e a degradação do tecido41 . AccBI1 e 2 são os genes mais expressos durante o amadurecimento das frutas, com leituras por kilobase por milhão de leituras mapeadas (RPKMs) chegando a 443.814. A expressão de AccBI1 oscila até 0 RPKMs em alguns estágios de maturação, sugerindo controle estrito da transcrição. O abacaxi F153 continha 61 genes de bromelina, incluindo dois que possuem padrões de expressão que se correlacionam inversamente com AccBI1 e AccBI2 (Fig. 6d ).
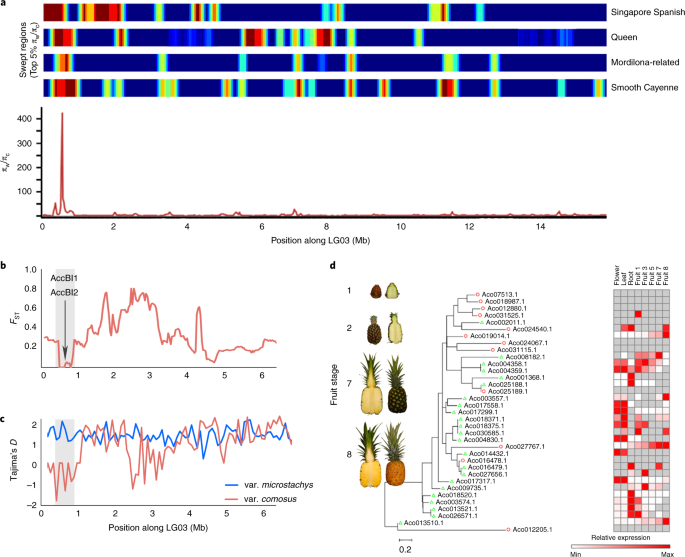
a , Top: Mapas de calor mostrando a distribuição de varreduras de domesticação (top 5% π w / π c ) para as quatro cultivares. Inferior: uma região varrida putativa no final de LG03 contendo AccBI1 e AccBI2 . π w / π c em todas as cultivares é plotado usando uma janela deslizante de 0,5 Mb com deslocamento de 0,1 Mb. b , Distância genética ( F ST ) entre os clusters relacionados a Smooth Cayenne, Queen, Singapore Singapore e Mordilona para os 6,5 Mb de LG03. Os valores médios de F ST são plotados em janelas deslizantes de 50 kb com tamanho de passo de 25 kb. c, Valores de D de Tajima para os quatro agrupamentos de cultivares combinados (var. Comosus ) e selvagem (var. Microstachys ). Os valores médios de D do Tajima são plotados em janelas deslizantes de 50 kb com um tamanho de passo de 25 kb. d , Esquerda: Frutos de abacaxi em estágios selecionados de uma série de maturação de frutos (estágios 1, 2, 7 e 8). Direita: Filogenia de probabilidade máxima de genes de bromelina com RPKMs de expressão transformada log 2 em frutos, flores, folhas e tecidos das raízes.
Genes candidatos à auto-incompatibilidade em abacaxi
Em contraste com A. macrodontes , A. comosus e suas variedades botânicas são auto-incompatíveis, com exceção de alguns clones de var. bracteatus . No entanto, a auto-incompatibilidade tende a ser mais forte na var. comosus , em comparação com as outras variedades que não foram domesticadas para frutas, o que provavelmente é resultado da seleção sob domesticação para reduzir a produção de sementes nos frutos 43 . Autoincompatibilidade gametofítica (GSI) operada em abacaxi cultivado 44 , semelhante ao GSI à base de S-RNase, no qual o locus S codifica uma única proteína S-RNase e várias proteínas F-box (SLFs / SFBs) 45. Quando autopolinizados em espécies SI, nenhum dos SLFs / SFBs interage com sua própria S-RNase, que decompõe o RNA do tubo de pólen para inibir o crescimento; quando polinizados cruzadamente, alguns membros de SLFs / SFBs paternos interagem com a S-RNase materna, o que permite o crescimento do tubo de pólen 45 . Para procurar genes potencialmente envolvidos no GSI de abacaxi, primeiro identificamos homólogos de S-RNase e SLF / SFB no genoma de referência de abacaxi com base na homologia de sequência. Esses candidatos foram testados quanto ao histórico de seleção em diversas variedades de abacaxi. Vinte e cinco genes passaram nos critérios (Tabela Suplementar 24 ).Examinamos os níveis de transcrição dos 25 genes candidatos a SI em androecium e gnoecium, respectivamente (Tabela Suplementar 24 ). Dois S -Os genes da RNase (Aco001100 e Aco004758), os potenciais determinantes da especificidade feminina em GSI, foram altamente expressos em ambos os tecidos, mas com expressão mais forte em androecium. Para os SLFs / SFBs, dois dos genes (Aco00868 e Aco011265) apresentaram expressão muito mais forte no androecium do que no gynoecium, enquanto dois deles (Aco015095 e Aco021447) apresentaram viés de expressão oposto. A expressão de quatro genes não foi detectada. Os 13 genes restantes mostraram expressão semelhante nos dois tecidos. Entre os seis genes que mostram expressão diferencial em androecium e gynoecium, o membro da família T2 da ribonuclease Aco001100 e o membro da família F-box Aco00868 estão intimamente ligados ao LG02, separados apenas a 1,8 Mb e são os candidatos mais prováveis à auto-incompatibilidade em A. comosus var. comosusCayenne liso F153. Além disso, o Aco001100 estava fortemente ligado a outros dois genes SLF / SFB (Aco001170 e Aco012216), uma característica do GSI 45 da RNase . No CB5, o membro da família da ribonuclease T2 CB5.v30014510 era o gene ortólogo de Aco001100 e vinculado a apenas um gene da família SLF / SFB (CB5.v30013780), que não é um sistema SI funcional (Tabela Suplementar 25 ).
Discussão
A montagem em nível cromossômico do genoma CB5 de abacaxi bracteatus lança mais luz sobre a biologia e evolução de Ananas.
Para superar o problema de montar um genoma heterozigoto, desenvolvemos
um algoritmo, Pseudohaplóide, que identifica e filtra contigs
heterozigotos, buscando sequências homólogas redundantes. Facilitados
pela tecnologia de sequenciamento de longa leitura, identificamos e
localizamos sequências mais repetitivas no genoma CB5, fornecendo
recursos abrangentes para estudar a evolução do genoma impulsionada
pelos TEs. Além disso, os pseudo-cromossomos desmontados no genoma F153
foram corrigidos com a assistência do genoma CB5. A comparação entre
esses dois genomas de abacaxi revelou componentes genômicos associados à
produção de fibras, formação de cor, acúmulo de açúcar e maturação dos
frutos. Também forneceu uma linha adicional de evidência para verificar
os genes candidatos a SI no F153.
Nossos dados genômicos indicaram a presença de um continuum de divergência, variando de baixa divergência entre grupos de cultivares modernas de abacaxi a divergência moderada entre taxa intimamente relacionados, como as variedades botânicas cultivadas comosus , bracteatus e erectifolius , até uma divergência muito maior na natureza. var. microstachys , que exibe valores de D xy normalmente observados em espécies recentemente divergentes 34 , 35 . Em contraste, F STrefletiu baixa diversidade nas principais cultivares, consistente com o gargalo da domesticação. A diversidade bastante reduzida nas cultivares, em relação ao progenitor selvagem, apontou para o grave gargalo de domesticação experimentado por essa cultura propagada por clonagem, e um excesso de alelos de frequência intermediária em dois grandes grupos de cultivares modernas indicou o potencial da propagação clonal para mascarar variantes deletérias recessivas em heterozigotos 46 .
A análise de mistura de cultivares de A. comosus revelou genótipos de mistura generalizados em 39 (44%) dos 89 acessos, detectados em todas as cultivares e variedades botânicas. Com relação aos processos evolutivos que operam durante a domesticação do abacaxi, nossos resultados indicaram um papel para a mistura antiga e recente e, portanto, a recombinação sexual e a seleção artificial subsequente na maioria das cultivares. Isso foi sustentado pela escassez de corridas terminais de homozigose ao longo dos cromossomos do abacaxi em duas das três principais cultivares. Isso indicou que tanto a recombinação sexual quanto mutações somáticas contribuíram para a diversidade fenotípica observada em Ananas. Parece que o verdadeiro grau de complexidade genômica do germoplasma usado em programas de melhoramento do século XX foi subestimado anteriormente.
Cultivares de abacaxi pré-colombianos iniciais foram selecionados por baixo teor de fibra de frutos e redução na produção de sementes por menor fertilidade e auto-incompatibilidade 47 . O genoma do abacaxi contém 25 varreduras seletivas, muito menos do que as de culturas sexualmente reprodutivas, como a 121 na soja 39 e a 186 no tomate 38 , apoiando a conclusão de uma mistura de seleção sexual e assexual do abacaxi. A varredura seletiva mais forte incluiu um par de inibidores de bromelina duplicados em tandemly, anteriormente sugeridos como importantes reguladores da senescência e amadurecimento de frutos de abacaxi neste fruto não climatérico 41 . A duplicação de genes é o motor da inovação evolutiva e tem sido associada a características de domesticação do tomate48 e framboesa preta 49 . O evento de duplicação do gene inibidor da bromelina provavelmente foi selecionado em variedades pré-colombianas.
Nossa hipótese inicial de trabalho era que as mutações somáticas eram a principal fonte de variação da domesticação do abacaxi. Nossos esforços para identificar varreduras seletivas mitóticas foram frutíferos no cultivar espanhol de Cingapura, como mostrado por extensas execuções terminais de homozigose, a marca registrada da seleção mitótica. No entanto, essa hipótese foi rejeitada para duas cultivares principais, Smooth Cayenne e Queen, embora corridas terminais esporádicas tenham sido detectadas em dois cromossomos, indicando reprodução clonal a longo prazo pontuada por reproduções sexuais. Meiose em abacaxi geralmente ocorre uma vez em cada 2 anos, enquanto que a recombinação em células mitóticas é contínua, mas em taxas muito baixas, cerca de 10 4 a 10 5 vezes menos frequente do que a recombinação meiótica 50 , 51. A uma frequência tão baixa e a natureza da produção clonal, apenas os eventos de recombinação mitótica que ocorreram no estágio de célula única dos tecidos reprodutivos, coroas, ventosas, escorregões e brotos, poderiam ser transmitidos às progênies e preservados para serem detectáveis. Além disso, uma recombinação sexual pode interromper execuções terminais de homologia que se formaram e foram mantidas por milhares de anos.
A hipótese da operação em uma etapa, em que a domesticação e a melhoria precoce são um resultado imediato de um único propagador clonal, podem ser responsáveis pela seleção de alguns clones de longa duração em algumas linhagens. Análises genômicas, particularmente aquelas que buscam execuções terminais de homozigose, podem ser aplicadas a outras culturas propagadas clonicamente para elucidar a extensão da recombinação sexual versus descendência vegetativa em sua história de domesticação. A coexistência de recombinação sexual e a operação em uma etapa entre diferentes cultivares podem ser comuns em culturas propagadas por clones. Algumas hipóteses controversas foram rejeitadas no passado, mas posteriormente validadas total ou parcialmente por novas tecnologias inovadoras ou resolução aprimorada de evidências, incluindo o 'domínio' versus 'superdominância'hipóteses para heterose e a teoria de Lamarck da herança das características adquiridas. A hipótese da operação de uma etapa para a domesticação de culturas propagadas por clones parece ser uma delas.
Nossos dados genômicos indicaram a presença de um continuum de divergência, variando de baixa divergência entre grupos de cultivares modernas de abacaxi a divergência moderada entre taxa intimamente relacionados, como as variedades botânicas cultivadas comosus , bracteatus e erectifolius , até uma divergência muito maior na natureza. var. microstachys , que exibe valores de D xy normalmente observados em espécies recentemente divergentes 34 , 35 . Em contraste, F STrefletiu baixa diversidade nas principais cultivares, consistente com o gargalo da domesticação. A diversidade bastante reduzida nas cultivares, em relação ao progenitor selvagem, apontou para o grave gargalo de domesticação experimentado por essa cultura propagada por clonagem, e um excesso de alelos de frequência intermediária em dois grandes grupos de cultivares modernas indicou o potencial da propagação clonal para mascarar variantes deletérias recessivas em heterozigotos 46 .
A análise de mistura de cultivares de A. comosus revelou genótipos de mistura generalizados em 39 (44%) dos 89 acessos, detectados em todas as cultivares e variedades botânicas. Com relação aos processos evolutivos que operam durante a domesticação do abacaxi, nossos resultados indicaram um papel para a mistura antiga e recente e, portanto, a recombinação sexual e a seleção artificial subsequente na maioria das cultivares. Isso foi sustentado pela escassez de corridas terminais de homozigose ao longo dos cromossomos do abacaxi em duas das três principais cultivares. Isso indicou que tanto a recombinação sexual quanto mutações somáticas contribuíram para a diversidade fenotípica observada em Ananas. Parece que o verdadeiro grau de complexidade genômica do germoplasma usado em programas de melhoramento do século XX foi subestimado anteriormente.
Cultivares de abacaxi pré-colombianos iniciais foram selecionados por baixo teor de fibra de frutos e redução na produção de sementes por menor fertilidade e auto-incompatibilidade 47 . O genoma do abacaxi contém 25 varreduras seletivas, muito menos do que as de culturas sexualmente reprodutivas, como a 121 na soja 39 e a 186 no tomate 38 , apoiando a conclusão de uma mistura de seleção sexual e assexual do abacaxi. A varredura seletiva mais forte incluiu um par de inibidores de bromelina duplicados em tandemly, anteriormente sugeridos como importantes reguladores da senescência e amadurecimento de frutos de abacaxi neste fruto não climatérico 41 . A duplicação de genes é o motor da inovação evolutiva e tem sido associada a características de domesticação do tomate48 e framboesa preta 49 . O evento de duplicação do gene inibidor da bromelina provavelmente foi selecionado em variedades pré-colombianas.
Nossa hipótese inicial de trabalho era que as mutações somáticas eram a principal fonte de variação da domesticação do abacaxi. Nossos esforços para identificar varreduras seletivas mitóticas foram frutíferos no cultivar espanhol de Cingapura, como mostrado por extensas execuções terminais de homozigose, a marca registrada da seleção mitótica. No entanto, essa hipótese foi rejeitada para duas cultivares principais, Smooth Cayenne e Queen, embora corridas terminais esporádicas tenham sido detectadas em dois cromossomos, indicando reprodução clonal a longo prazo pontuada por reproduções sexuais. Meiose em abacaxi geralmente ocorre uma vez em cada 2 anos, enquanto que a recombinação em células mitóticas é contínua, mas em taxas muito baixas, cerca de 10 4 a 10 5 vezes menos frequente do que a recombinação meiótica 50 , 51. A uma frequência tão baixa e a natureza da produção clonal, apenas os eventos de recombinação mitótica que ocorreram no estágio de célula única dos tecidos reprodutivos, coroas, ventosas, escorregões e brotos, poderiam ser transmitidos às progênies e preservados para serem detectáveis. Além disso, uma recombinação sexual pode interromper execuções terminais de homologia que se formaram e foram mantidas por milhares de anos.
A hipótese da operação em uma etapa, em que a domesticação e a melhoria precoce são um resultado imediato de um único propagador clonal, podem ser responsáveis pela seleção de alguns clones de longa duração em algumas linhagens. Análises genômicas, particularmente aquelas que buscam execuções terminais de homozigose, podem ser aplicadas a outras culturas propagadas clonicamente para elucidar a extensão da recombinação sexual versus descendência vegetativa em sua história de domesticação. A coexistência de recombinação sexual e a operação em uma etapa entre diferentes cultivares podem ser comuns em culturas propagadas por clones. Algumas hipóteses controversas foram rejeitadas no passado, mas posteriormente validadas total ou parcialmente por novas tecnologias inovadoras ou resolução aprimorada de evidências, incluindo o 'domínio' versus 'superdominância'hipóteses para heterose e a teoria de Lamarck da herança das características adquiridas. A hipótese da operação de uma etapa para a domesticação de culturas propagadas por clones parece ser uma delas.
Métodos
Visão geral da montagem e anotação do genoma
A montagem no nível cromossômico CB5 aproveita a tecnologia em tempo real PacBio de molécula única e os métodos de andaimes baseados em Hi-C, seguidos pelo polimento baseado em leitura curta da Illumina. Resumidamente, ~ 50 × a cobertura de sub-leituras foi gerada com a plataforma PacBio RSII e ~ 60 × a cobertura por leituras curtas foi gerada na plataforma Illumina HiSeq X10. A montagem inicial no nível de contig foi realizada com CANU v.1.7 e contigs heterozigotos foram removidos usando nosso algoritmo recém-desenvolvido, Pseudohaplóide (detalhes em Nota Complementar) Além disso, recrutaram-se leituras curtas de Illumina para polir o genoma montado PacBio usando o Pilon v.1.18 com os parâmetros: - diplóide - roscas 6 - alterações - trilhas - bases fixas - verbose - mindepth 4. As bibliotecas Hi-C foram criadas a partir de folhas macias do CB5 na BioMarker Technologies Corporation, conforme descrito anteriormente 52 . Os fragmentos quiméricos que representam os fragmentos reticulados originais foram então processados em bibliotecas de sequenciação de extremidade pareada e sequenciados na plataforma Illumina HiSeq X10. As leituras das extremidades emparelhadas foram mapeadas de forma exclusiva no conjunto de rascunho e contigs desassociados foram corrigidos através da detecção de padrões abruptos de contato de longo alcance usando o pipeline 3D-DNA 53 . Os contigs corrigidos pelo Hi-C foram ainda ligados em 25 pseudo-cromossomos usando o pipeline ALLHiC 54 .Anotamos o conjunto de nível cromossômico do CB5 usando uma série de programas, que são totalmente descritos na Nota Complementar . Resumidamente, o pipeline MAKER2 55 foi usado para anotar as proteínas codificadoras de proteínas integrando proteínas homólogas, transcritos montados em RNA-seq e os resultados de preditores de genes ab initio. Além disso, as sequências repetitivas foram previstas pelo RepeatMasker 56 e também previmos miRNAs pesquisando candidatos que correspondessem aos miRNAs públicos.
Identificação dos genes biossintéticos da lignina e da antocianina
As sequências de proteínas de genes da biossíntese da lenhina de Arabidopsis thaliana , Populus trichocarpa e Oryza sativa 20 foram usadas para alinhar com as sequências de proteínas de F153 e CB5 usando BLASTP com um ponto de corte e o valor ≤ 1 × 10 -10 e cobertura ≥ 0,75. Para identificação dos genes biossintéticos da antocianina, sequências proteicas de Arabidopsis thaliana 57foram utilizados para alinhar com as sequências proteicas de F153 e CB5. O Pfam foi adotado para identificar domínios conservados para esses genes candidatos. Finalmente, usamos o MEGA 7 para desenhar uma árvore filogenética para confirmar a relação esperada dos genes biossintéticos da antocianina. A árvore filogenética foi inferida pelo método de união de vizinhos. O alinhamento foi feito pelo MUSCLE v.3.8.31 com modelo de substituição padrão e 1.000 autoinicializações.Identificação dos genes da subfamília C1 da CP
Os modelos genéticos de todas as espécies utilizadas neste estudo foram baixados do Phytozome v.11.0 e v.12.0 ( https://phytozome.jgi.doe.gov/pz/portal.html ). O domínio conservado da subfamília C1 de cisteína peptidase, domínio peptidase_C1 (PF00112) foi baixado do banco de dados pfam ( http://pfam.xfam.org/ ). O HMMER foi usado para pesquisar bancos de dados de proteínas de cada espécie para identificar proteínas contendo o domínio peptidase_C1 com limiar de valor e ≤ 1 × 10-5. Confirmamos ainda essas proteínas pesquisando seus domínios no Banco de Dados de Domínios Conservados do NCBI. As proteínas de comprimento total foram alinhadas pelo MUSCLE v.3.8.31 com os parâmetros padrão. As árvores filogenéticas foram construídas pelo Smart Model Selection PhyML v.3.0 com critério estatístico (AIC) 58 e posteriormente editadas com o MEGA 7.Chamada e anotação de variantes
Um total de 4,7 bilhões de leituras de Illumina com extremidade emparelhada de 150–250 bp rendeu uma cobertura média de 17,5 × por adesão (Tabela Suplementar 19 ). Essa profundidade de leitura é semelhante a outros projetos de sequenciamento em larga escala 39 , 59 , 60 . As leituras brutas foram filtradas com qualidade para remover adaptadores e bases de baixa qualidade (Q <30 abacaxi="" alinhadas="" as="" bowtie2="" com="" de="" f153="" filtrada="" font="" foram="" genoma="" leituras="" mascarado="" n="" o="" qualidade="" ref.="" usando="" v.2.2.6="" v.6="">61 ) com parâmetros padrão. As taxas de leitura de mapeamento para adesões cultivadas variou de 82.3% a 94.5% com uma média de 87,6% em comparação com 69,4-84,2% para selvagem Ananase espécies relacionadas. A detecção de variantes foi realizada usando o kit de ferramentas de análise do genoma (GATK; v.3.5-0-g36282e4) 62seguindo o fluxo de trabalho de práticas recomendadas para descoberta de variantes. Os arquivos BAM resultantes foram realinhados localmente usando o IndelRealigner para remover incompatibilidades incorretas em torno de inserções e exclusões em pequena escala. As variantes foram chamadas em cada acesso separadamente usando o HaplotypeCaller e os arquivos de formato de chamada de variante do genoma individual (gVCF) foram mesclados usando GenotypeGVCFs. Essa abordagem em duas etapas inclui recalibração e regenotipagem de qualidade no arquivo vcf mesclado, garantindo a precisão da variante. O sinalizador –output_mode EMIT_ALL_CONFIDENT_SITES foi usado para fornecer cobertura de leitura para cada posição no genoma de referência (incluindo sites invariantes), permitindo que regiões sem alinhamento sejam filtradas antes da análise genética da população. Um total de 9.342.943 variantes brutas foram chamadas pelo GATK.Essas variantes foram filtradas para remover sites com índices de qualidade inferiores a 100, frequência mínima de alelos <0 ausentes="" dados="" e=""> 10%. O arquivo vcf final contém 7.428.400 SNPs e indels de alta qualidade (<10 89="" acessos.="" anotadas="" as="" font="" foram="" nos="" pb="" ref.="" snpeff="" usando="" v.4.2="" variantes="">63 ) com modelos genéticos de abacaxi 19 .Análise de frequência de alelos de sites não-sinônimo e sinônimo
Sites não-sinônimo e sinônimo anotados pelo SNPEff foram usados para análise de frequência de alelos do site. Somente SNPs de acessos espanhóis Smooth Cayenne, Queen e Cingapura foram utilizados devido ao seu maior tamanho de amostra. A variedade botânica de bracteatus foi usada como um grupo externo para polarizar variantes ancestrais e derivadas. A frequência do alelo foi estimada separadamente para cada população e foram analisadas as posições do SNP em mais de 70% do tamanho da amostra de cada população. Como cada posição do SNP tinha tamanhos de amostra diferentes, usamos a distribuição hipergeométrica para amostragem reduzida do tamanho de amostra observado na j- ésima posição do SNP, N j , até o tamanho mínimo amostral reduzido da amostra em todas as posições do SNP, n (ref. 64) Assim, a frequência do alelo para um tamanho de amostra inferior de n foi calculada como:Análise de desequilíbrio de ligação (LD)
O arquivo vcf final foi usado para o cálculo da LD em todo o genoma, usando indivíduos com histórico evolutivo não misturado (Fig. 3c ). Usando PLINK (v.1.90b3.46) (ref. 65 ), o LD entre pares de SNP dentro do mesmo LG foi calculado usando uma janela de 5 Mb e limitando a SNPs que não tinham mais de 499.999 SNPs separados. Foram analisados SNPs dentro de LGs com pelo menos 10 Mb de comprimento. Os pares SNP foram então agrupados em caixas de 10 kb para calcular a média da correlação ao quadrado R ( r 2 ) entre os SNPs. Pares SNP com valores de r 2 <0 font="" foram="" omitidos.="">O método LOESS da linha de melhor ajuste foi ajustado usando o valor médio de r 2 por caixa.Análise RNA-seq
As leituras emparelhadas ou de extremidade única aparadas de cada amostra foram alinhadas ao genoma F153 com máscara repetida v.6 (ref. 19 ), usando TopHat (v.2.0.9) nas configurações padrão 66 . O valor RPKM normalizado de cada amostra foi estimado pelo Cufflinks v.2.2.1, seguido pelo Cuffnorm v.2.2.1 (ref. 66 ) usando configurações padrão com a anotação do modelo de gene do abacaxi (v.6) 19 .Análise de mistura, filogenética e estrutura populacional
Os SNPs da nova sequenciação de genoma foram filtrados usando vcftools v.0.1.13 (ref. 67 ) com contagem alélica mínima = 2, dados perdidos máximos = 15%, cobertura mínima = 4, qualidade SNP> 20, mantendo apenas variantes bialélicas e nenhuma indels. Uma árvore com base em probabilidade máxima de acessos a Ananas foi construída usando RAxML v.8.2 (ref. 28 ) com 100 réplicas de autoinicialização para determinar o suporte a ramificações, e uma rede filogenética foi construída usando o método de rede vizinha implementado no SplitsTree 68 . Além disso, o MDS foi usado para agrupar sem acessos de modelo os acessos de Ananas . Diversidades de nucleotídeos ( π ), D xy e F STforam estimados para todos os táxons e principais cultivares. Diversidades de nucleotídeos nas formas selvagens e cultivadas foram usadas como uma abordagem simples e robusta para documentar gargalos genéticos experimentados durante a domesticação; evitamos a modelagem demográfica da história das cultivares usando abordagens baseadas em difusão ou coalescentes devido à presença generalizada de genótipos propagados por clonagem no conjunto de amostras, o que violaria as premissas básicas de modelagem. Em vez disso, exploramos os principais aspectos da história das cultivares, analisando os padrões genômicos de ancestralidade. A ancestralidade e a mistura do genoma foram estimadas com o ADMIXTURE v.1.23 (ref. 69 ). Para variedade comosusPara as cultivares, essa análise utilizou apenas as cinco amostras com maior cobertura para cada cultivar, a fim de evitar vieses devido à super-representação de amostras clonais. As divisões da população e os eventos de mistura anteriores foram ainda mais explorados usando a abordagem TreeMix 70 . A ancestralidade local ao longo dos cromossomos de abacaxi confiantemente montados foi estimada com uma abordagem do Modelo de Markov Escondido modificado por Price et al. 71 seguindo Wegmann et al. 72 , utilizando o software RASPberry. O número mais provável de gerações desde a mistura foi estimado para cada indivíduo misturado por esse método, com base em testes de razão de verossimilhança. Exceto quando indicado, análises estatísticas foram realizadas em R.Detectando varreduras seletivas putativas
Regiões de seleção durante a domesticação de abacaxi foram identificadas com base em reduções drásticas em π de acessos cultivados em comparação com linhas selvagens ( π c / π w ) em janelas deslizantes em todo o genoma. As microstáquias das variedades são o provável progenitor do abacaxi cultivado; portanto, a diversidade nesse grupo foi usada para estimar π w. As cultivares com evidência de mistura foram omitidas nas varreduras de seleção e π c foi calculado dentro e através de cada uma das quatro cultivares. Para reduzir os falsos positivos devido à deriva, os quatro grupos cultivados foram combinados em um único pool antes da análise. Diversidade de nucleotídeos ( π) foi calculado usando a ferramenta –window-pi-step no vcftools (v.0.1.12) (ref. 67 ). Locais invariantes foram incluídos nos cálculos de π para remover quaisquer inflações nas estimativas relacionadas à falta de dados. A diversidade de nucleotídeos foi calculada em janelas deslizantes de 50 kb com um tamanho de 10 kb para identificar varreduras e em janelas deslizantes de 10 kb com um passo de 2,5 kb para restringir genes candidatos. Os 5% superiores dos valores de π w / π c foram considerados regiões varridas. As janelas varridas adjacentes foram mescladas em blocos, produzindo um conjunto final de 25 regiões varridas.As regiões varridas por candidatos foram mais estreitadas usando uma abordagem baseada em XP-CLR para modelar as diferenças no espectro de frequências alélicas entre acessos cultivados e selvagens 73 . Os seguintes parâmetros foram usados para varreduras XP-CLR em cada cromossomo: janela de 0,005 cM, tamanho de janela de 1.000 pb, máximo de 100 SNPs por grade e um nível de correção de 0,7. A distância genética entre variantes adjacentes foi calculada usando o mapa genético de alta densidade usado para ancorar o genoma de abacaxi F153 19 . Comparações foram feitas entre var. cultivares de comosus que não mostram evidência de mistura recente e var. microstachys . As regiões com as melhores pontuações de XP-CLR de 10% foram mescladas como regiões de varredura putativas e apenas regiões que se sobrepõem a altasOs valores de π w / π c foram mantidos para remover os falsos positivos.
A ST foi estimada com a abordagem Weir e Cockerman usando comparações de quatro vias dos agrupamentos de cultivares (Smooth Cayenne, Queen, Singapore Spanish e Mordilona) no programa SFselect ( https://github.com/rronen/SFselect ). O D de Tajima foi calculado em janelas deslizantes de 50 kb com sobreposição de 25 kb usando um conjunto de programas em vcftools (v.0.1.12) 67 .
Identificação e mapeamento de locais de inserção de elementos transponíveis
O MITE-Hunter 74 foi usado com parâmetros padrão para pesquisar o conjunto do genoma do abacaxi em busca de pequenos EEs de DNA. Os resultados do MITE-Hunter foram examinados manualmente para selecionar EEs de boa-fé com base em suas seqüências de flanqueamento, características de TIR e TSD e classificados em famílias seguindo a convenção usada por Han et al. 75. As 50 bases terminais de TEs foram usadas como consultas por explosão para identificar junções TE no genoma do abacaxi. Os resultados da explosão foram filtrados para reter hits com comprimento mínimo de alinhamento de 15 pb e estão a 10 pb dos terminais TE. Múltiplos hits de explosão dentro de uma janela de 30 pb foram mesclados e considerados como uma junção. Esses hits marcam junções TE exclusivas no genoma do abacaxi de referência. A presença / ausência de junções TE foi pontuada nos 89 acessos com base no mapeamento das leituras Illumina desde os acessos até o genoma de abacaxi de referência. Um local foi marcado como presente em uma adesão quando pelo menos uma leitura abrangeu 20 pb a montante e a jusante da junção TE.Identificando trilhas de homozigotia
As faixas de homozigotia foram identificadas utilizando as 50 variedades ressequenciadas com a maior cobertura. Longos tratos de homozigose são geralmente regiões genômicas com genes consecutivos sem heterozigose. Identificamos primeiro todos os tratos de homozigose que abrangem mais de três genes consecutivos. Nos raros casos em que dois tratos de homozigose foram interrompidos por apenas um gene com apenas um SNP heterozigótico, as três partes ainda estavam unidas em tratos mais longos de homozigose. Em seguida, foi contado o número de tratos de homozigose que abrangem seis ou mais genes consecutivos e foi apresentado o somatório dos números de tratos de homozigose entre as 38 cultivares, com 100 genes em tamanho de lixeira. Linhas pontilhadas vermelhas marcam as localizações previstas dos centrômeros,com base na observação de que, de longe, a maior densidade de retrotransposons LTR sempre flanqueia o centrômero em todos os genomas de angiospermas estudados18 .Resumo dos relatórios
Informações adicionais sobre o design da pesquisa estão disponíveis no Resumo dos Relatórios de Pesquisa da Natureza vinculado a este artigo.Disponibilidade de dados
O genoma e a anotação do bracteatus CB5, a versão revisada do genoma F153 e os dados de RNA-seq de abacaxi foram submetidos à EBI-ENA sob o estudo PRJEB33121 . As
leituras filtradas de qualidade da Illumina para os 89 genomas de
abacaxi sequestrados foram depositadas no banco de dados do NCBI
BioProject ( http://www.ncbi.nlm.nih.gov/bioproject ) sob o número de acesso PRJNA389669 .
Disponibilidade de código
Desenvolvemos
um novo algoritmo, Pseudohaplóide, que identifica e filtra contigs
heterozigotos com base no alinhamento de todo o genoma. Esse método pode ser executado de forma independente com qualquer montador e está disponível de código aberto em http://github.com/schatzlab/pseudohaploid . Outros
softwares / códigos de código aberto e personalizados disponíveis ao
público, usados para analisar os dados deste estudo, estão listados no
Nature Research Reporting Summary .
Referências
- 1 Zohary, D. Seleção inconsciente e evolução das plantas domesticadas. Econ. Robô. 58 , 5-10 (2004).
- 2) Bertoni, MS Contribuições para o Estudo Botânico das Plantas Cultivadas (Ex Sylvis. Puerto Bertoni, Alto Paraná. PY, 1919).
- 3) Byers, DS Pré-História do Vale Tehuacan (Univ. Of Texas Press, 1967).
- 4) Coppens d'Eeckenbrugge, G. e Duval, M.‑F. A domesticação do abacaxi: contexto e hipóteses. Pineapple News 16 , 15–27 (2009).
- 5) Coppens d'Eeckenbrugge, G., Uriza Ávila, DE, Rebolledo Martínez, A. & Rebolledo Martínez, L. O Bloco Cascajal: outro testemunho da antiguidade do abacaxi no México? Pineapple News , 18 , 47-48 (2011).
- 6 Baker, KF & Collins, JL Notas sobre a distribuição e ecologia de Ananas e Pseudananas . Sou. J. Bot. 26 , 697-702 (1939).
- 7) Duval, MF, Coppens d'Eeckenbrugge, G., Ferreira, FR, Bianchetti, LDB & Cabral, JRS Primeiros resultados da coleta conjunta de germoplasma EMBRAPA-CIRAD Ananas no Brasil e na Guiana Francesa. Acta Hortic. 425 , 137-144 (1997).
- 8) Asim, M. et al. Uma revisão sobre o abacaxi deixa a fibra e seus compósitos. Int. J. Polym. Sci . 2015 , 950567 (2015).
- 9 Beltrame, KK et al. Adsorção de cafeína em fibras de carvão ativado mesoporoso preparadas a partir de folhas de abacaxi. Ecotox. Environ. Seguro. 147 , 64–71 (2018).
- 10) Abd Razak, SI, Sharif, NFA, Nayan, NHM, Muhamad, II e Yahya, MY Impregnação de poli (ácido láctico) em fibras de folhas de abacaxi com polpa biologicamente orgânica para materiais de embalagem. Bioresources 10 , 4350-4359 (2015).
- 11) Costa, LMM et al. Bionanocompósitos de extrato de casca de PVA / nanofibras de abacaxi / Stryphnodendron adstringens eletrospun para aplicações médicas. Ind. Crop. Prod. 41 , 198-202 (2013).
- 12) Hazarika, P. et al. Desenvolvimento de aparelhos a partir de resíduos de seda e fibra de folhas de abacaxi. J. Nat. Fibers 15 , 416-424 (2018).
- 13) Benzing, DH Bromeliaceae: perfil de uma radiação adaptativa . (Cambridge Univ. Press, 2000).
- 14) Givnish, TJ et al. Radiação adaptativa, evolução correlacionada e contingente e diversificação líquida de espécies em Bromeliaceae. Mol. Phylogenet. Evol. 71 , 55-78 (2014).
- 15 Barbará, T., Martinelli, G., Fay, M., Mayo, S. e Lexer, C. Diferenciação populacional e coesão de espécies em duas plantas intimamente relacionadas, adaptadas aos inselbergs neotropicais de alta altitude, Alcantarea imperialis e Alcantarea geniculata ( Bromeliaceae). Mol. Ecol. 16 , 1981-1992 (2007).
- 16 Wendt, T., Canela, MBF, de Faria, APG e Rios, RI Biologia reprodutiva e hibridação natural entre duas espécies endêmicas de Pitcairnia (Bromeliaceae). Sou. J. Bot. 88 , 1760-1767 (2001).
- 17 Palma-Silva, C. et al. Espécies de bromélias simpáticas ( Pitcairnia spp.) Facilitam testes de mecanismos envolvidos na coesão e isolamento reprodutivo de espécies em inselbergs neotropicais. Mol. Ecol. 20 , 3185–3201 (2011).
- 18 Bennetzen, JL & Wang, H. As contribuições de elementos transponíveis para a estrutura, função e evolução dos genomas vegetais. Annu. Rev. Plant Biol. 65 , 505-530 (2014).
- 19 Ming, R. et ai. O genoma do abacaxi e a evolução da fotossíntese CAM. Nat. Genet. 47 , 1435-1442 (2015).
- 20 Hamberger, B. et ai. Análises em todo o genoma de genes relacionados ao fenilpropanóide em Populus trichocarpa , Arabidopsis thaliana e Oryza sativa : a caixa de ferramentas Populus lignin e conservação e diversificação de famílias de genes da angiosperma. Pode. J. Bot. 85 , 1182–201 (2007).
- 21 Ehlting, J. et al. O perfil global de transcrição de hastes primárias de Arabidopsis thaliana identifica genes candidatos para elos ausentes na biossíntese de lignina e reguladores transcricionais da diferenciação de fibras. Plant J. 42 , 618-640 (2005).
- 22) Adisak, J. & Jintana, J. no VII International Pineapple Symposium vol. 902 (eds Abdullah, H. et al.) 423-426 (Sociedade Internacional de Ciências Hortícolas, 2011).
- 23 Jiang, SY et al. Famílias de genes do metabolismo da sacarose e suas funções biológicas. Sci. Rep. 5 , 17583 (2015).
- 24 Ruan, YL Metabolismo da sacarose: porta de entrada para uso diverso de carbono e sinalização de açúcar. Ann. Rev. Plant Biol. 65 , 33-67 (2014).
- 25) Büttner, M. A família do gene transportador de monossacarídeos (semelhante a) em Arabidopsis . Cartas FEBS 581 , 2318-2324 (2007).
- 26) Doidy, J. et ai. Transportadores de açúcar nas plantas e em suas interações com fungos. Tendências Plant Sci. 17 , 413-422 (2012).
- 27 Coppens d'Eeckenbrugge, G., Sanewski, GM, Smith, MK, Duval, M.-F. & Leal, F. in Wild Crop Paratives: Genomic and Breeding Resources (ed. Kole C.) 21-41 (Springer, 2011).
- 28 Stamatakis, A. RAxML-VI-HPC: análises filogenéticas baseadas em máxima verossimilhança com milhares de táxons e modelos mistos. Bioinformatics 22 , 2688-2690 (2006).
- 29 Huson, DH SplitsTree: analisando e visualizando dados evolutivos. Bioinformatics 14 , 68-73 (1998).
- 30) Coppens d'Eeckenbrugge, G., Duval, M.-F., Leal, F. in Genetics and Genomics of Pineapple (ed. Ming, R.) 1–25 (Springer, 2018).
- 31 Chapman, MA, Hiscock, SJ e Filatov, DA Divergência genômica durante especiação impulsionada pela adaptação à altitude. Mol. Biol. Evol. 30 , 2553–2567 (2013).
- 32 Cruickshank, TE & Hahn, MW Reanalysis sugere que as ilhas genômicas de especiação são devidas à diversidade reduzida, e não ao fluxo gênico reduzido. Mol. Ecol. 23 , 3133-3157 (2014).
- 33 Eckert, CG em Ecologia e Biologia Evolutiva de Plantas Clonais (eds Stuefer, JF et al.) 279-298 (Springer, 2002).
- 34) Chen, J., Hu, Q., Zhang, Y., Lu, C. e Kuang, H. P-MITE: um banco de dados para elementos transponíveis de repetição invertida em miniatura de plantas. Nucleic Acids Res. 42 , D1176-D1181 (2014).
- 35) Wessler, SR, Bureau, TE & White, LTR-retrotransposons e MITEs: atores importantes na evolução dos genomas vegetais. Curr. Opin. Genet. Dev. 5 , 814-821 (1995).
- 36 Stern, C. Travessia somática e segregação em Drosophila melanogaster . Genetics 21 , 625 (1936).
- 37) LaFave, MC & Sekelsky, J. Recombinação mitótica: por que? quando? quão? Onde? PLoS Genet. 5 , e1000411 (2009).
- 38 Lin, T. et ai. As análises genômicas fornecem insights sobre a história da criação de tomate. Nat. Genet. 46 , 1220–1226 (2014).
- 39 Zhou, Z. et al. Resequenciar 302 acessos silvestres e cultivados identifica genes relacionados à domesticação e melhoria da soja. Nature Biotechnology 33 , 408-414 (2015).
- 40 Ramu, P. et ai. HapMap de mandioca: Gerenciando a carga genética em uma espécie de cultura clonal. Pré- impressão em bioRxiv https://doi.org/10.1101/077123 (2016).
- 41 Neuteboom, LW, Matsumoto, KO e Christopher, DA Um tronco N-terminal rico em AE estendido em cistatina de abacaxi secretada aumenta a inibição da bromelina da fruta e é removido após a tradução durante o amadurecimento. Plant Physiol. 151 , 515-527 (2009).
- 42 Raimbault, AK, Zuily-Fodil, Y., Soler, A., Mora, P. e de Carvalho, MHC Os padrões de expressão da bromelina e do AcCYS1 correlacionam-se com a resistência do coração negro em frutos de abacaxi submetidos ao estresse pós-colheita. J. Plant Physiol. 170 , 1442-1446 (2013).
- 43 Coppens d'Eeckenbrugge, G., Duval, M.‑F. & Van Miegroet, F. Fertilidade e auto-incompatibilidade no gênero Ananas . Acta Hortic. 334 , 45-52 (1992).
- 44) Brewbaker, JL & Gorrez, DD Genética da auto-incompatibilidade nos gêneros monocotiledôneas, Ananás (abacaxi) e Gasteria . Sou. J. Bot. 54 , 611-616 (1967).
- 45 Bedinger, PA, Broz, AK, Tovar-Mendez, A. & McClure, B. Interações pólen-pistilo e seu papel na seleção de parceiros. Plant Physiol. 173 , 79-90 (2017).
- 46 Gaut, BS, Seymour, DK, Liu, QP e Zhou, YF Demografia e seus efeitos na variação genômica na domesticação de culturas. Nat. Plants 4 , 512-520 (2018).
- 47 Coppens d'Eeckenbrugge, G., Leal, F. & Bartholomew, D. em The Pineapple: Botany, Production and Uses 13–32 (2003).
- 48) Xiao, H., Jiang, N., Schaffner, E., Stockinger, EJ e van der Knaap, E. Uma duplicação gênica mediada por retrotransposon está subjacente à variação morfológica do tomate. Science 319 , 1527–1530 (2008).
- 49 VanBuren, R. et ai. O genoma da framboesa preta ( Rubus occidentalis ). Plant J. 87 , 535-547 (2016).
- 50 Paques, F. & Haber, JE Várias vias de recombinação induzidas por quebras de fita dupla em Saccharomyces cerevisiae . Microbiol. Mol. Biol. Rev. 63 , 349-440 (1999).
- 51 Petes TD & Symington LS em Biologia Molecular e Celular dos Leveduras Saccharomyces (eds Jones, EW et al.) 407-521 (Cold Spring Harbor Press, 1991).
- 52 Xie, T. et ai. Montagem do genoma vegetal de novo com base nas interações da cromatina: um estudo de caso de Arabidopsis thaliana . Mol. Plant 8 , 489-492 (2015).
- 53 Dudchenko, O. et al. A montagem de novo do genoma de Aedes aegypti usando Hi-C produz andaimes com comprimento de cromossomo. Science 356 , 92–95 (2017).
- 54 Zhang, J. et al.Genoma definido pela alelo da cana-de- açúcar autopoliplóide Saccharum spontaneum L. Nat. Genet. 50 , 1565-1573 (2018).
- 55 Campbell, MS, Holt, C., Moore, B. e Yandell, M. Anotação e Curadoria do Genoma Usando MAKER e MAKER-P. Curr. Protoc. Bioinformtics 48 , 11–39 (2014).
- 56
- 57 Guo, N. et al. Genes biossintéticos da antocianina em Brassica rapa . BMC Genomics 15 , 426 (2014).
- 58 Lefort, V., Longueville, J.‑E. & Gascuel, O. SMS: seleção inteligente de modelos em PhyML. Mol. Biol. Evol. 34 , 2422-2424 (2017).
- 59 Hazzouri, KM et al. O sequenciamento completo do genoma das tamareiras produz insights sobre a diversificação de uma cultura de árvores frutíferas. Nat. Comum. 6 , 8824 (2015).
- 60 Qi, J. et ai. Um mapa de variação genômica fornece insights sobre a base genética da domesticação e diversidade de pepinos. Nat. Genet. 45 , 1510-1515 (2013).
- 61 Langmead, B. & Salzberg, SL Alinhamento rápido de leitura de brechas com Bowtie 2. Nat. Métodos 9 , 357–359 (2012).
- 62 DePristo, MA et al. Uma estrutura para descoberta de variações e genotipagem usando dados de sequenciamento de DNA de última geração. Nat. Genet. 43 , 491-498 (2011).
- 63 Cingolani, P. et al. Um programa para anotar e prever os efeitos de polimorfismos de nucleotídeo único, SnpEff: SNPs no genoma da Drosophila melanogaster cepa w1118; iso-2; iso-3. Voar (Austin). 6 , 80-92 (2012).
- 64 Nielsen, R. et ai. Uma varredura de genes selecionados positivamente nos genomas de humanos e chimpanzés. PLoS Biol. 3 , 170 (2005).
- 65) Purcell, S. et ai. PLINK: um conjunto de ferramentas para a associação de genoma inteiro e análises de ligação baseadas na população. Sou. J. Hum. Genet. 81 , 559-575 (2007).
- 66 Trapnell, C. et al. Análise diferencial de expressão gênica e de transcrição de experimentos de RNA-seq com TopHat e Abotoaduras. Nat. Protoc. 7 , 562-578 (2012).
- 67 Danecek, P. et al. O formato de chamada variante e VCFtools. Bioinformatics 27 , 2156-2158 (2011).
- 68 Huson, DH & Bryant, D. Aplicação de redes filogenéticas em estudos evolutivos. Mol. Biol. Evol. 23 , 254–267 (2006).
- 69 Alexander, DH, Novembre, J. & Lange, K. Estimativa rápida baseada em modelo de ancestralidade em indivíduos não relacionados. Genome Res. 19 , 1655-1664 (2009).
- 70 Pickrell, JK & Pritchard, JK Inferência de divisões populacionais e misturas de dados de frequência de alelos em todo o genoma. PLoS Genet. 8 , e1002967 (2012).
- 71 Price, AL et al. Detecção sensível de segmentos cromossômicos de ancestralidade distinta em populações misturadas. PLoS Genet. 5 , e1000519 (2009).
- 72 Wegmann, D. et ai. Taxas de recombinação em indivíduos misturados identificados por inferência ancestral. Nat. Genet. 43 , 847-53 (2011).
- 73 Chen, H., Patterson, N. & Reich, D. Diferenciação populacional como um teste para varreduras seletivas. Genome Res. 20 , 393-402 (2010).
- 74 Han, Y. & Wessler, SR MITE-Hunter: um programa para descobrir elementos transponíveis de repetição invertida em miniatura a partir de seqüências genômicas. Nucleic Acids Res. 38 , e199 (2010).
- 75 Han, Y., Qin, S. & Wessler, SR A comparação de elementos transponíveis de classe 2 na resolução de superfamília revela características conservadas e distintas nos genomas da grama de cereais. BMC Genomics 14 , 71 (2013).
Reconhecimentos
Este
trabalho foi financiado pelo Departamento de Ciência e Tecnologia da
província de Fujian (concessão nº 2016NZ0001-1), a Fundação Nacional de
Ciências Naturais da China (nos. U1605212 para YQ, 31628013 para QY e
31701874 para XZ), a Fuzhou Science e projeto de tecnologia (concessão
2017-N-33 a XZ), o Fundo Distinto para Jovens Bolsistas da Universidade
de Agricultura e Florestas de Fujian (concessão n ° xjq201609 ao LYC), a
National Science Foundation concede (nos. DBI-1401572 a RV e
DBI-1350041 para MCS), Institutos Nacionais de Saúde dos EUA (concessão
nº R01-HG006677 para MCS), do Genoma Vegetal NSF (nos. 0607123 e
043707-01 para JLB e IOS-1546218 para MDP), Fundação Zegar Family
Concessão (no. A16-0051-001 ao MDP) e doação da Swiss National Science
Foundation (no. CRSII3_147630 ao CL). Agradecemos a W. Till, D. Wegmann e
D.Bartholomew para discussões úteis e J. Lin para assistência na
submissão de dados.
Informação sobre o autor
Notas do autor
Afiliações
Contribuições
A RM concebeu esse projeto genoma e coordenou atividades de pesquisa. RM, LYC, RV e JLB projetaram os experimentos. TM, PB, QW e M.-LW mantiveram e forneceram germoplasma de Ananas . O MCS e o XZ desenvolveram algoritmos para resolver montagens redundantes. XZ, HY, SC, MA, SR, MCS, RV e CMW processaram dados de sequenciamento e sequenciamento de genomas. LYC, XZ, Z.Liao, J.Liu, J.Lin, JY, MF, Z.Lin, JZ, LH, J.Wu, SZ, LW, YQ, T.-YH, S.-MK, C. -WT, M.-LW e REP analisaram o genoma de CB5 e 'F153' e os dados de RNA-seq. Analisaram-se os genomas equivalentes RV, MP, HZ, CMW, JZ, LH, HW, JYC, AS, JS, WCY, JCC, JW, QY, GCdE, GMS, MDP, JLB, CL e RM. RV, RM, LYC, XZ, GCdE, GMS, CL e JLB escreveram o manuscrito.Autores correspondentes
Correspondências para Jeffrey L. Bennetzen ou Christian Lexer ou Ray Ming .Declarações de ética
Interesses competitivos
Os autores declaram não ter interesses concorrentes.Informação adicional
Nota do editor A Springer Nature permanece neutra em relação a reivindicações jurisdicionais em mapas publicados e afiliações institucionais.
Informação suplementar
Informação suplementar
Nota complementar e figuras suplementares 1–24 e tabelas suplementares 1–28.
Conjunto de dados suplementar 1
Lista de genes de biossíntese de lignina em Arabidopsis thaliana , Populus trichocarpa , Oryza sativa e Ananas comosus.
Nenhum comentário:
Postar um comentário
Observação: somente um membro deste blog pode postar um comentário.