The study of ancient DNA is
hampered by degradation, resulting in short DNA fragments. Advances in
laboratory methods have made it possible to retrieve short DNA
fragments, thereby improving access to DNA preserved in highly degraded,
ancient material. However, such material contains large amounts of
microbial contamination in addition to DNA fragments from the ancient
organism. The resulting mixture of sequences constitutes a challenge for
computational analysis, since microbial sequences are hard to
distinguish from the ancient sequences of interest, especially when they
are short.
Results
Here, we develop a method to
quantify spurious alignments based on the presence or absence of rare
variants. We find that spurious alignments are enriched for mismatches
and insertion/deletion differences and lack substitution patterns
typical of ancient DNA. The impact of spurious alignments can be reduced
by filtering on these features and by imposing a sample-specific
minimum length cutoff. We apply this approach to sequences from four
~ 430,000-year-old Sima de los Huesos hominin remains, which contain
particularly short DNA fragments, and increase the amount of usable
sequence data by 17–150%. This allows us to place a third specimen from
the site on the Neandertal lineage.
Conclusions
Our method maximizes the
sequence data amenable to genetic analysis from highly degraded ancient
material and avoids pitfalls that are associated with the analysis of
ultra-short DNA sequences.
After its death, the DNA of an organism inevitably degrades into short DNA fragments [1, 2]. Laboratory methods have been developed that specifically aim at retrieving these fragments from ancient biological material [3, 4, 5] and transforming them efficiently into library molecules for high-throughput sequencing [6].
These developments have enabled researchers to study DNA sequences from
increasingly older samples. One notable example are four remains from
Sima de los Huesos in Spain that constitute, with an age of over
400,000 years, the by far oldest hominin material to date that yielded
ancient DNA sequences [7, 8].
Owing to their great age, the vast majority of hominin DNA fragments
that can be extracted from the Sima de los Huesos remains are shorter
than 45 bp [7].
In
addition to the extreme state of DNA fragmentation, the analysis of
sequences from highly degraded material is hampered by the large number
of extraneous DNA fragments originating from microorganisms that
decomposed the remains of the source organism after its death [9, 10, 11, 12]. In the case of Sima de los Huesos [8]
and many other ancient skeletal remains, microbial DNA constitutes more
than 99% of the DNA that can be recovered and sequenced. Contaminant
sequences are typically differentiated from those that stem from the
source organism by aligning all sequences to a related reference genome
and retaining only those that produce alignments with not more than a
pre-defined number of differences [13, 14].
However, unrelated sequences can align by chance and the probability of
such spurious alignments increases with decreasing sequence length [15]. This issue is expected to affect particularly the analysis of sequences from highly fragmented material.
To
minimize the effect of spuriously aligning sequences on downstream
analyses, previous studies employed sequence length cutoffs that have
been gauged by a variety of methods. Green et al. [16]
used specific alignment software to analyze the distribution of
alignment scores at various sequence lengths. This distribution was
found to be distinctly bimodal at longer lengths, as expected from a
mixture of related and unrelated sequence alignments, while bimodality
was not observed at shorter lengths. Setting a length cutoff that
preserves the bimodal distribution can thus be used to limit the
fraction of spurious alignments.
Cutoffs have also been determined by
testing at which lengths mammoth sequences yielded equally good
alignments to other mammalian taxa [10, 13], horse sequences aligned equally well to the chicken genome [17], mammoth and ancient bovine sequences aligned to a database of concatenated bacterial genomes [18], or fragmented bacterial genomes aligned to the human reference [19].
While these methods have been sufficient to determine approximate
cutoffs, they do not provide an estimate of the fraction of spurious
alignments. We also note that microbial genomes in public databases may
present a poor proxy for the microbial sequence diversity found in real
sequence data from ancient remains. The validity of these approaches is
therefore hard to judge.
More recently, Meyer et al. [8]
used a different approach to determine sequence length cutoffs for the
analysis of nuclear DNA sequences from the Sima de los Huesos samples.
Using sequence variants that are unique to the human reference genome,
as determined by comparison to known variation from human resequencing
studies and the genomes of non-human primates, they counted the fraction
of sequences that match the reference-specific variant. These variants
are rare and are expected to be largely absent in other hominin genomes.
In contrast, spuriously aligned sequences will match the reference
genome by chance, independent of how frequent the reference genomes’
variants are in the human population. Since no matches to the
reference-specific variant was observed for sequences of at least 35-bp
length, this cutoff was deemed sufficient to exclude spurious
alignments. However, due to the limited number of unique reference
variants (i.e., 11,299) and the small amount of data obtained from the
Sima de los Huesos remains (less than 0.001-fold genomic coverage per
sample), only between 4 and 69 sequences formed the basis for this
assessment, preventing any fine-scale estimates of the fraction of
spurious alignments.
Here,
we test and extend this approach to allow for the confident estimation
of the fraction of spurious alignments across different sequence
lengths. We use these estimates to devise sequence length cutoffs that
maximize the number of useful sequences and increase the power of
phylogenetic analysis. Applying our approach to the Sima de los Huesos
samples, we determine that cutoffs shorter than 35 bp are suitable for
some of these samples, as long as appropriate filters are put in place.
The increase in usable sequences allows us to confidently place one of
the Sima de los Huesos samples on the Neandertal lineage that previously
yielded inconclusive results.
To
allow for fine-scale estimates of the fraction of spurious alignments
in small datasets, we changed ~ 18 million interspersed bases in the
human reference genome (see the “Methods”
section). These artificial mutations were introduced at positions where
the human reference, all human genomes sequenced as part of the 1000
Genomes project, two high-coverage archaic human genomes, and the
chimpanzee genome show the same base. They are thus unlikely to occur in
present-day or ancient hominin genomes (probability < 0.1%).
Spurious alignments, on the other hand, are likely to match the mutated
state (Fig. 1a). The alignment parameters used here and in other studies [14, 20] limit the fraction of allowed mismatches per alignment to approximately 10% (see the “Methods”
section), resulting for spuriously aligned sequences in a predicted
~ 90% match probability for the mutated state and a ~ 3.3% probability
for matching either of the remaining three states (Fig. 1a).
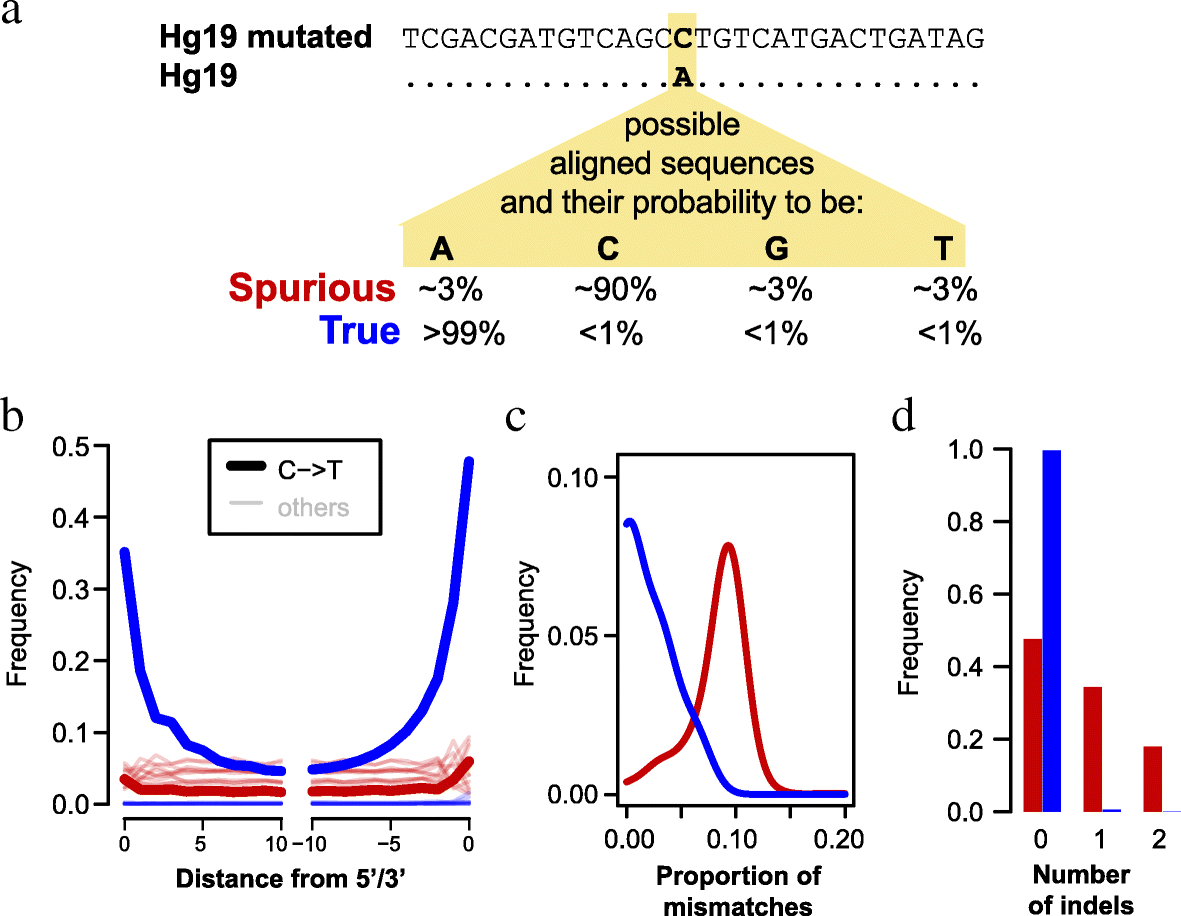
Fig. 1
Identification and characterization of spurious and true sequence alignments. a
Schematic illustration of how spurious and true sequence alignments are
inferred. The human reference (hg19) is mutated to introduce changes at
positions that are not known to vary among present-day humans and other
hominins. True hominin sequences (blue) and spuriously aligned
microbial sequences (red) are expected to show the reference, the
mutated or one of the two other states with the probabilities indicated.
b Frequency of all nucleotide substitutions at each position in Mezmaiskaya 1 sequence alignments. c
Distributions of the proportion of mismatches in Mezmaiskaya 1
alignments. One mismatch was subtracted from all true alignments and
those spurious alignments that did not carry the mutated allele. This
was done to compensate for the fact that these alignments have to carry a
mismatch to the mutated reference genome in order to be identified as
such. d Distributions of the number of indels in Mezmaiskaya 1 alignments. See Additional file 1:
Figure S1 for the distribution of mismatches and indels with the modern
human and the bacterial datasets that were used as negative and
positive controls, respectively
To
test whether these predictions hold, we generated sequences from DNA
isolated from the blood sample of a healthy human individual that was
fragmented heavily to mimic the size distribution of ancient DNA. We
further compiled a dataset consisting of 3860 bacterial genomes that
were cut in silico into 3.03 billion unique sequences uniformly
distributed between 20- and 40-bp length (see the “Methods”
section). We then mapped both sets of sequences to the mutated
reference and counted the fraction of sequences that match the reference
state at mutated positions (presumed hominin alignment, henceforth
“true alignment”) or any other variant (presumed “spurious alignment”).
Of the aligned human sequences, 99.8% were correctly classified as
hominin. Out of 782 million bacterial sequences that could be aligned to
the mutated reference, 97.6% were correctly classified as spurious
(Additional file 1:
Table S1). If all alignments of bacterial sequences contained the
maximal number of allowed mismatches, 3.9% of the sequences would be
expected to carry the reference state by chance, whereas we observe a
lower percentage of 2.4% (see Additional file 1:
Tables S2 and S3 for a similar analysis with cut sequences from a
protist and a fungus genome). Since the percentage of misclassified
sequences biases the estimated fraction of spurious alignments slightly
downward, we corrected our estimates in all subsequent analyses using
conservatively the expected proportion (see the “Methods” section).
Characteristics of spurious and true alignments
We
next investigated whether spurious and true alignments differ in
specific characteristics. For this purpose, we aligned sequences from
the Mezmaiskaya 1 Neandertal [20, 21], a published dataset containing a considerable fraction of ultra-short (< 35 bp) sequences (Additional file 1: Figure S2) and approximately 9% Neandertal DNA, to the mutated reference. After filtering for mappability (see the “Methods”
section) and classifying the alignments as described above, we obtained
5.07 million true alignments and 0.92 million spurious alignments. The
procedure uses strand orientation to avoid misclassifying sequences due
to ancient DNA damage (see the “Methods” section).
We
first note that true Mezmaiskaya 1 sequence alignments show elevated
frequencies of C-to-T substitutions, which occur predominantly at their
beginning and ends (Fig. 1b).
This pattern is expected for authentic ancient DNA sequences and
results from deamination of cytosine to uracil in single-stranded DNA
overhangs [22].
In contrast, this pattern is not observed for spurious alignments,
where C-to-T substitutions are similar in frequency to other types of
substitutions. Second, we find that true alignments carry significantly
fewer mismatches on average than spurious alignments (0.018 vs 0.108 per
bp; Wilcoxon rank sum test p value < 2.2e−16; see Fig. 1c).
The fraction of mismatches in the true alignments is still
substantially larger than the genomic divergence between modern humans
and Neandertals of < 0.002 differences per base pair [21]. However, C-to-T substitutions account for most of this difference (Fig. 1b).
Third, true alignments contain fewer insertions/deletions (indels) than
spurious alignments (0.5% vs. 52.4% of the alignments, Wilcoxon rank
sum test, p value < 2.2e−16) (Fig. 1d). Indels accumulate at a roughly 10 times lower rate than single nucleotide mutations in humans [23] and are therefore expected to be rare in true alignments.
We repeated these analyses using the bacterial and modern human control datasets (Additional file 1: Figure S1 and Table S4). Similar to the results from spurious Mezmaiskaya 1 alignments (Fig. 1c, d),
bacterial alignments are enriched for mismatches (0.092 per bp on
average) and indels (76.2% of the alignments), whereas mismatches and
indels are rare among modern human control alignments (0.004 per bp and
0.05%, respectively).
Minimizing the proportion of spurious alignments
We
next binned all Mezmaiskaya 1 sequences by length and calculated the
fraction of spurious alignments for each bin. As expected, the fraction
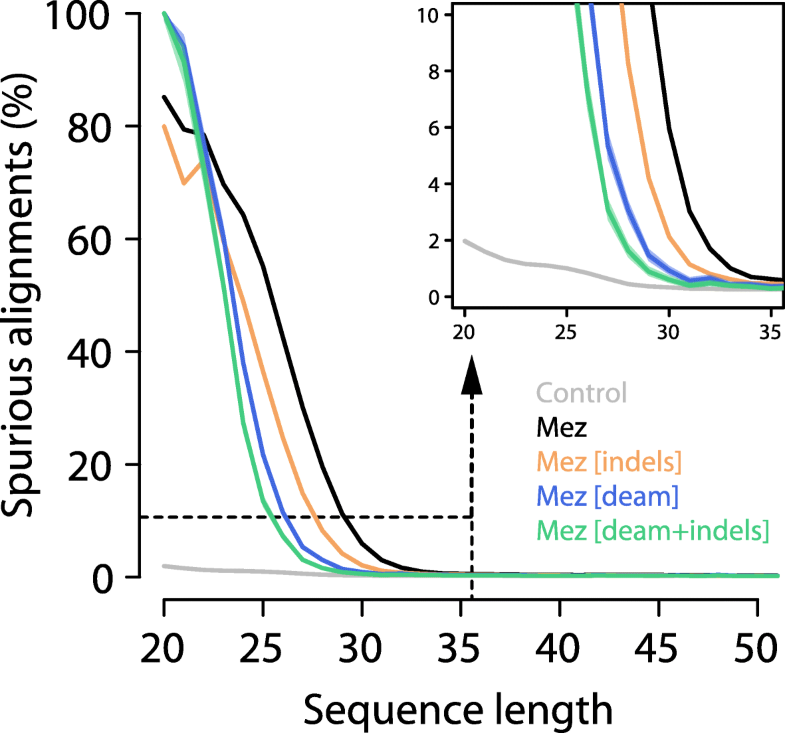
of spurious alignments increases with decreasing sequence length (Fig. 2).
Spurious alignments are rare (< 0.3%) in sequences of at least 35 bp
length, suggesting that a sequence length cutoff of 35 bp, which was
used in several ancient DNA studies (Additional file 1:
Table S5), is effective in removing the vast majority of spurious
alignments for the Mezmaiskaya 1 dataset analyzed here. In fact, even
sequences of length 33 bp show a proportion of spurious alignments of
less than 1%, indicating that shorter sequences could be included in
downstream analyses (Fig. 2).
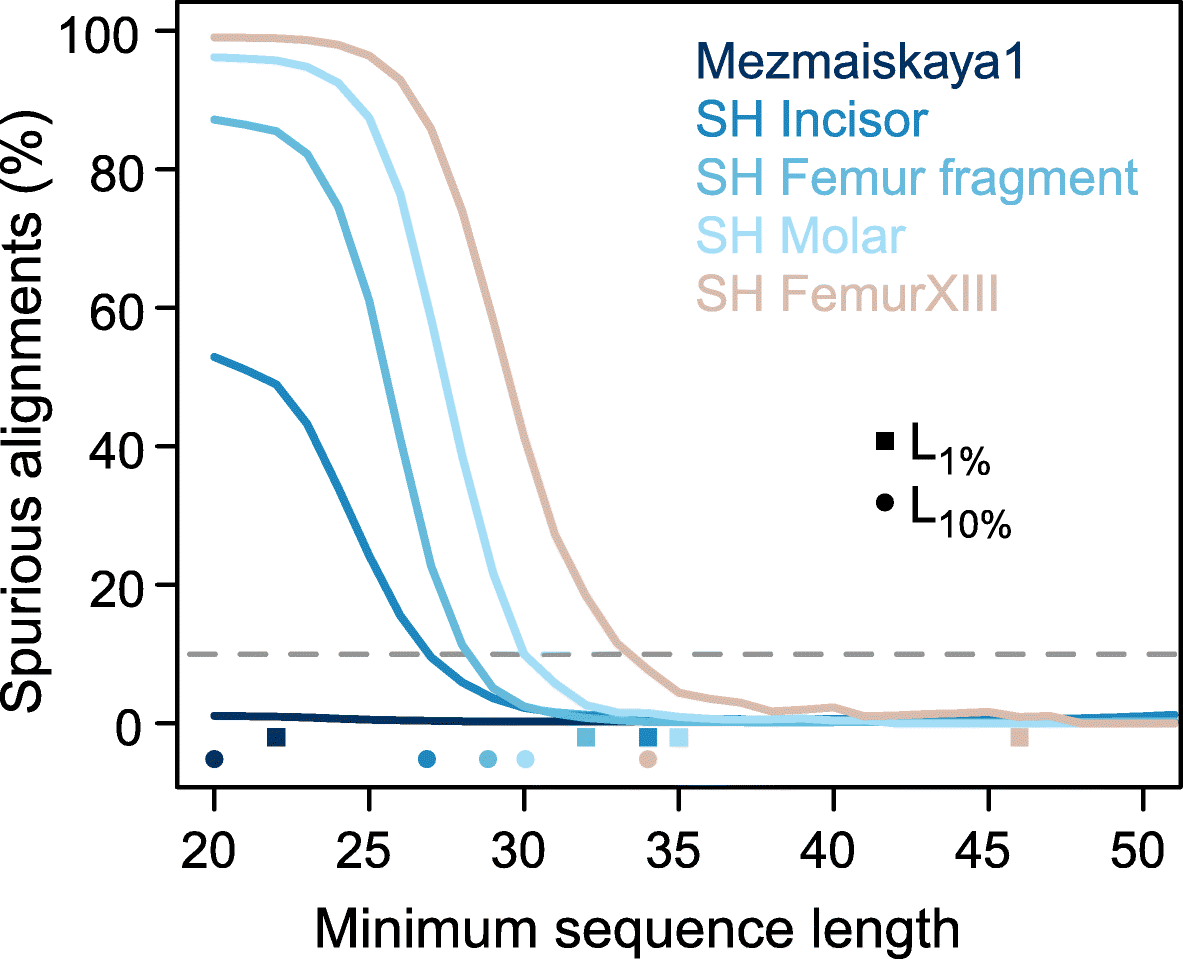
Fig. 2
Effect of
sequence length, indel, and deamination filters on the proportion of
spurious alignments. The proportion of spurious alignments in each size
bin is shown for all Mezmaiskaya 1 (“Mez”) alignments in black,
alignments without indels in orange, alignments with terminal C to T
substitutions (“deam”) in blue, and with both filters applied in green.
An analysis of modern human sequences (“Control”) without microbial
contamination is shown in gray. Ninety-five percent binomial confidence
intervals are as wide as or smaller than the line width. The dashed
rectangle encloses the area depicted in the zoom-in on the top-right
corner
The
previous analysis has shown that spurious alignments lack the elevation
of terminal C-to-T substitution frequencies that are typical for
ancient DNA and that they contain more indels than true alignments
(Fig. 1).
Filtering based on these features may thus help to further reduce the
fraction of spurious alignments. In agreement with this assumption, we
find that restricting the analysis to alignments exhibiting a C-to-T
substitution at either terminus yields less than 1% spurious alignments
for length bins as short as 30 bp. It should be noted that this
deamination filter is often used to deplete sequence data of human
contamination. However, it also removes a large fraction of potentially
genuine ancient sequences that were not affected by deamination (~ 85%
of aligned sequences ≥ 35 bp in Mezmaiskaya 1). A less pronounced effect
is observed when removing alignments with indels (~ 1% of aligned
sequences ≥ 35 bp in Mezmaiskaya 1), which yields less than 1% spurious
alignments in size bins of 32 bp or longer. Combining both filters
reduces this number to 29 bp. The reduction of spurious alignments
achieved with both filters is also reflected by a decrease in sequence
differences to the reference genome (Fig. 2).
We
repeated this analysis using our modern human control sample, which
should, by design, not produce any spurious alignments. We find that
even the shortest length bin yields an estimate for the proportion of
spurious alignments of less than 2% (Fig. 2), suggesting that sequencing or mapping errors have little impact on our measure.
A re-analysis of sequences from Sima de los Huesos
The
extremely short DNA sequences that have been retrieved from the Sima de
los Huesos remains are an ideal dataset to explore to which extent the
choice of sequence filters changes the amount of useful sequence data
that can be obtained from very poorly preserved material and the
inferences that can be drawn from these data. Appreciable amounts of
nuclear DNA sequences are available from four hominin remains from the
site [8];
the fraction of hominin DNA varies between 0.02 and 0.18% in these
samples when considering sequences of at least 35-bp length. However,
the vast majority (> 97%) of the human aligned sequences of at least
20-bp length are shorter than this 35-bp cutoff (Additional file 1: Figure S2).
To
determine whether at least some of these ultra-short sequences are
amenable to analysis, we realigned the data of all four samples to the
mutated reference genome and removed alignments that contained indels
and those showing no evidence of deamination (see also Additional file 1:
Figures S3 and S4). The deamination filter is strictly required when
working with these data, as a substantial fraction of the hominin
sequences is derived from modern human contamination [7, 8].
We then calculated sequence length cutoffs that limit the fraction of
spurious alignments to < 1% or < 10%, henceforth denoted by L1% and L10%, respectively.
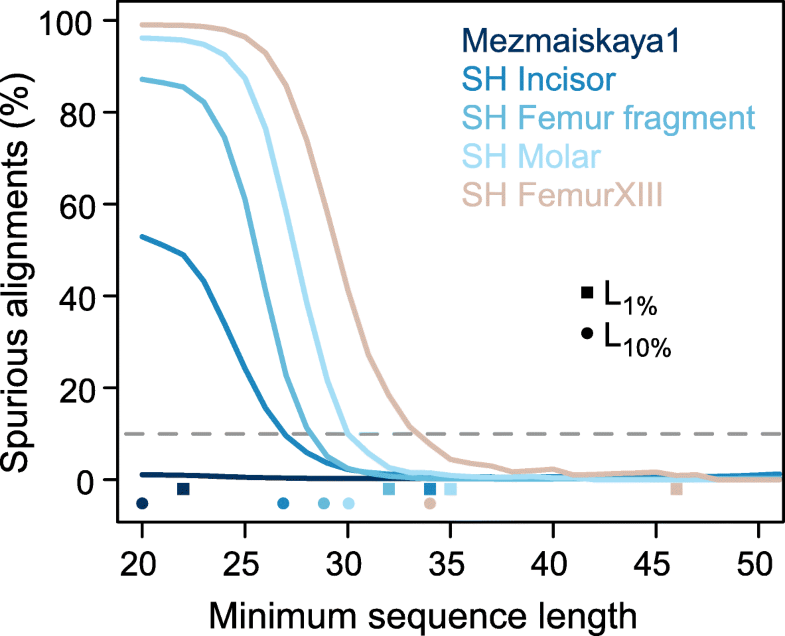
The four samples yield L10% cutoffs that range from 27 to 34 bp and decrease with increasing proportions of endogenous DNA (Fig. 3, Table 1).
Applying these cutoffs instead of the previously used cutoff of 35 bp
would increase the usable data by 17–150%. The more conservative L1% cutoffs would result in 0–40% more data for three of the four samples. Interestingly, the fourth sample, FemurXIII, yields a L1%
cutoff of 46 bp, suggesting that the often applied cutoff of 35 bp is
not always sufficient to limit spurious alignments to low levels. In
comparison, sequences from the Mezmaiskaya 1 Neandertal yield an L1% of 22 bp and do not reach a limit for L10%
(less than 10% of all sequences of at least 20-bp length aligned
spuriously). Considering sequences of at least 20 bp for analysis would
result in 37% more data compared to a 35-bp length cutoff.
Fig. 3
Cumulative proportion of spurious alignments. Squares and dots on the x-axis show the length-cutoffs that guarantee a spurious alignment rate lower than 1% (L1%) and lower than 10% (L10%), respectively (see also Table 1).
The dashed horizontal gray line indicates 10% spurious alignments. Only
sequences with C-to-T changes in the terminal 5′ and 3′ positions and
without indels are considered (i.e., the filters “deam+indels” used in
Fig. 2)
Table 1
Mezmaiskaya and Sima de los Huesos (SH) sequence length cutoffs allowing for less than 1% or 10% spurious alignments
Samples
Hominin DNA (%)a
Length cutoff (bp)b
Total number of hominin bases recovered (Mbp)
Fold change
L1%
L10%
35 bp
L1%
L10%
L1%/35 bp
L10%/35 bp
Mezmaiskaya 1
8.84
22
20
88.53
121.17
121.73
1.37
1.37
SH Femur frag.
0.11
32
29
0.81
1.14
1.52
1.41
1.88
SH Incisor
0.18
34
27
1.49
1.70
3.71
1.14
2.49
SH Molar
0.03
35
30
0.13
0.13
0.25
1.00
1.98
SH FemurXIII
0.02
46
34
0.15
0.03
0.18
0.23
1.17
aThe
percentage of endogenous hominin DNA was calculated as the fraction of
sequences of at least 35 bp that mapped to the human reference over the
total number of sequences
bThe values L1% and L10% refer to length cutoffs that limit the fraction of spurious alignment to under 1% and 10%, respectively. Column 35 bp
refers to the standard 35-bp length threshold. Values have been
computed using sequences with terminal C-to-T changes only and
disregarding sequences with indels
Since
present-day human contamination constitutes a challenge for the
analysis of archaic human sequences, we also tested whether
contamination rates differ when including shorter sequences. We found no
significant differences in the estimated contamination compared to the
previously used length cutoff of 35 bp, although this result may be
caused by a lack of power for the Sima de los Huesos samples
(Additional file 1: Table S6). We note that contamination estimates tend to be higher using L10%
cutoffs likely due to a reference bias, causing spurious alignments to
match the human reference allele more likely than the archaic allele.
We
also note that non-human eukaryotic contaminants would not be expected
to be enriched among shorter sequences since contaminant sequences tend
to be longer and the reference bias acts against their alignment [24].
Improving phylogenetic inferences from limited data
The
initial analysis of nuclear DNA sequences from the Sima de los Huesos
specimens revealed that two of the specimens (an incisor and a femur
fragment) share significantly more derived alleles with the
high-coverage genome of a Neandertal than with that of a Denisovan
individual [8].
While this result concurred with the fact that the Middle Pleistocene
Sima de los Huesos remains were discovered in the western part of the
territory inhabited by Neandertals during the Late Pleistocene (Europe
and Central Asia), it deviated from the mitochondrial tree [7],
which groups the Sima de los Huesos hominins into a clade with
Denisovans, who are thought to have inhabited large parts of Asia [25, 26].
To
test whether the inclusion of data from shorter nuclear sequences would
affect inferences about the phylogenetic position of the Sima de los
Huesos specimens, we compared the results of the lineage assignment test
(see the “Methods” section) obtained by using a 35-bp cutoff, as previously published, and the L10%
cutoffs determined here for all four specimens for which at least 1000
sequences from putatively deaminated DNA fragments were available (Fig. 4).
For the femur fragment and the incisor, the inclusion of additional
data strengthens the confidence of the Neandertal lineage assignment,
and the significance of the assignment was highest when between 2.5 and
15.6% of spurious alignments were allowed (Additional file 1:
Figure S5). This suggests that a spurious alignment proportion of
around 10% can be tolerated for this analysis. We caution that such a
high proportion of spurious alignments is not necessarily tolerable by
other types of analysis and that similar tests need to be carried out to
determine appropriate cutoffs.
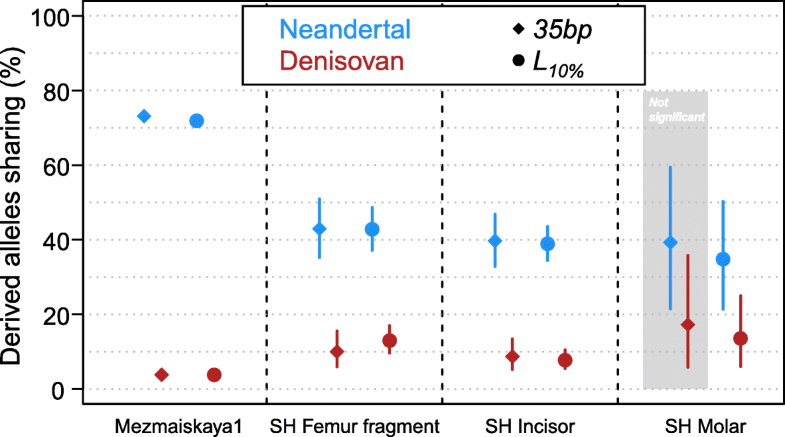
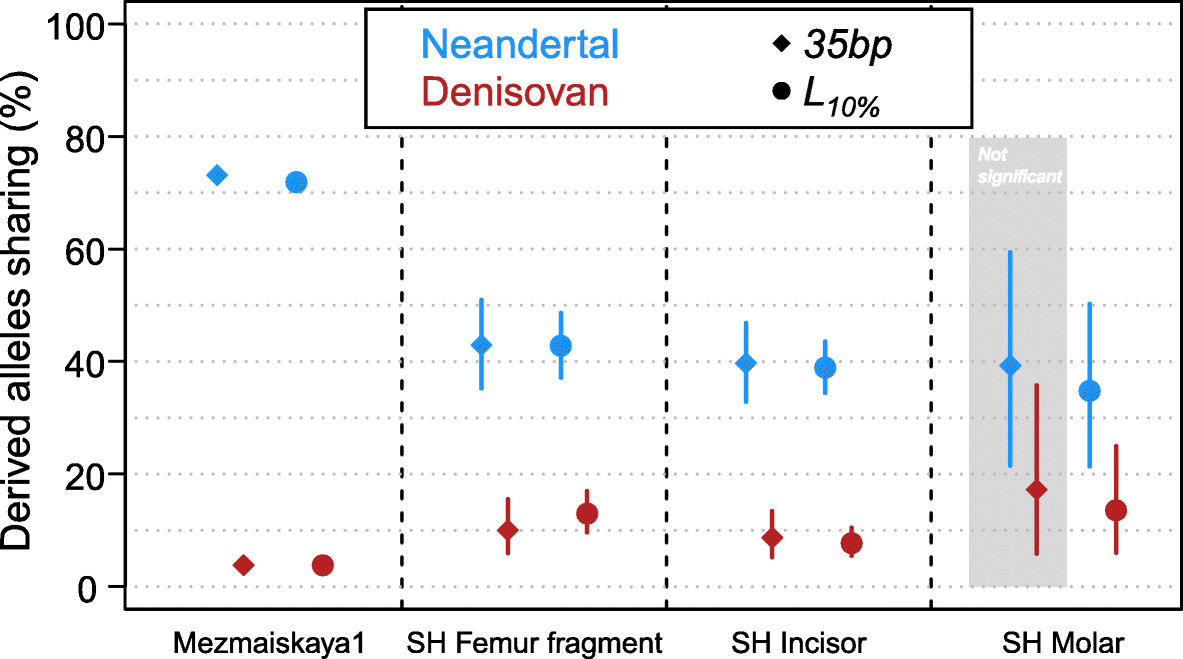
Fig. 4
Percentage of derived allele sharing with the Denisovan and Neandertal lineages. The circles and diamonds correspond to the L10% and 35-bp
length cutoffs, respectively. Bars indicate 90% binomial confidence
intervals. The difference between Neandertal and Denisovan sharing is
statistically significant in all comparisons, except for the SH Molar
with the 35-bp cutoff highlighted in the gray area (Fisher exact test p value = 0.09)
As
previously, one of the other Sima de los Huesos samples (Femur XIII)
did not yield sufficient data for a confident lineage assignment and no
additional data could be gained by applying the L10%
cutoff. However, the fourth specimen, a molar, shows significantly
higher allele sharing with the Neandertal than the Denisovan genome with
the L10% cutoff (Fig. 4, Fisher’s exact test p value = 0.005 corrected for multiple testing [27]).
Moreover, the percentage of Neandertal-shared derived alleles of the
molar (35%) does not significantly differ from the percentages observed
for the incisor and the femur fragment (43% and 39%, respectively; all
pairwise Fisher’s exact tests p values > 0.29).
Phylogenetic inferences and reference bias
Since
the fraction of mismatches in alignments is limited, spuriously
aligning sequences are expected to exhibit a strong bias towards showing
the human reference allele. This preference for the human reference
allele should introduce a bias towards supporting the modern human
lineage in the lineage assignment analysis of spurious alignments. In
agreement with this expectation, we observe a strong bias towards the
human reference allele in misaligning bacterial sequences, which are
assigned to the modern human lineage (~ 33% of the human derived
variants shared). A similar signal is also observed for the lineage
assignment of Sima de los Huesos when considering size cutoffs that are
expected to lead to an overwhelming majority of spurious alignments
(Additional file 1: Figure S6). While our results with the L10%
cutoffs do not show significant differences to previous, more
conservative cutoffs for these samples, we caution that reference bias
may affect analyses and needs to be considered before including a higher
fraction of spurious alignments.
Experimental procedures have made great strides forward in extracting short ancient DNA fragments [3, 5, 6].
However, the resulting short sequences constitute a challenge for
computational processing since unrelated and related sequences cannot
easily be distinguished. This has led to the paradoxical situation, in
which short DNA fragments that are preserved in highly degraded samples
can be made accessible to sequencing, only to be discarded in downstream
computational analyses to avoid spurious alignments.
How
can shorter sequences be made available for analysis without increasing
the fraction of spurious alignments unduly? We have shown here that one
answer lies in specific filters that enrich for genuine alignments. By
filtering for sequences with evidence for deamination and without
insertion/deletion differences to the reference genome, we were able to
reduce the fraction of spurious alignments sufficiently to allow for the
inclusion of sequences shorter than 35 bp from three Sima de los Huesos
samples in phylogenetic analysis. This analysis confirmed that two of
the samples originate from early Neandertals and enabled us to place one
additional sample, a molar, on the Neandertal lineage. The Neandertal
allele sharing of this sample is similar to that of the other two. All
three samples could thus originate from a single group of early
Neandertal ancestors or relatives thereof.
The
highly degraded remains from Sima de los Huesos yielded, arguably, the
most challenging dataset in ancient DNA to date, containing a large
fraction of ultra-short sequences and a large fraction of sequences from
microbial contamination. In light of these difficulties, it is
encouraging for future work on material with poor DNA preservation that
useful genetic information could be recovered from ultra-short sequences
of three samples from the site. However, we have to acknowledge that
working with such sequences remains a challenge. Perhaps the best
example of this is given by our analysis of a fourth Sima de los Huesos
sample, FemurXIII, for which a minimum sequence length cutoff of 46 bp
must be applied to ensure that the fraction of spurious alignments is
restricted to less than 1%. This result shows that microbial
contamination is so abundant in this sample that the commonly used
cutoffs of 35 bp length or shorter (Additional file 1:
Table S5) is insufficient to reduce the effect of spurious alignments
to conservative levels. As more data from highly degraded material
become available, it will be crucial to ensure that spurious alignments
are quantified to avoid false results.
On
a broader level, our results show that the genetic analysis of poorly
preserved ancient biological material is limited not only by our ability
to extract and sequence the DNA it may contain, but also by our ability
to distinguish sequences that are endogenous to the organism from the
overwhelming majority of microbial contamination. Molecular methods have
been developed in the past to decrease the fraction of microbial
contamination. These methods used restriction enzymes that cut motifs
occurring preferentially in contaminant DNA [16], enriched for endogenous DNA fragments via hybridization capture [28] or depleted contaminant DNA prior to DNA extraction [29, 30]. Further research will be needed to establish how these methods can contribute to the study of highly degraded samples.
We
conclude that while spurious alignments are an inevitable issue for the
analysis of short ancient sequences, their influence can be accurately
assessed and limited by appropriate filtering. Together with further
refinement of molecular methods our approach paves the way towards the
study of older or more degraded samples.
The
human reference genome (hg19/GRCh37) was used as a template to create a
genome with additional single nucleotide changes. These changes were
introduced in conserved regions where the reference human base is
identical to the aligned bases of the chimpanzee pantro4 genome, the
high-coverage genomes of the Altai Neandertal [20] and the Denisovan [6], 24 high-coverage modern human genomes [6, 20], and all 2504 modern human individuals of the 1000 Genomes Project data phase 3 [31].
Sites 5-bp up- and downstream of all indels detected in these datasets
were excluded. Sites were also required to fall outside of simple
repeats annotated using the Tandem Repeat Finder [32] and to overlap positions of unique mappability based on 35mers [20].
Bases were changed every 100 bp. If a change fell in a region that was
excluded, the closest included position was determined and chosen as new
location if it was at least 75 bp from the closest adjacent changed
site. Bases were replaced by other bases according to probabilities that
keep the overall nucleotide composition identical to that of the hg19
genome. A total of 18,002,060 sites were modified.
Sequence data and alignments to the modified reference
We
used one lane of Illumina HiSeq 2500 sequencing data from the
Mezmaiskaya1 Neandertal individual (library R5661; see Suppl. 2 in ref. [21]) and the published sequences from Sima de los Huesos samples [8] Femur fragment, Incisor, Molar, and FemurXIII. Both datasets were generated with the same extraction method [3] and the single stranded DNA library protocol [33].
As
negative control—i.e., as a sample for which we do not expect to see
any spurious alignments—we used modern human DNA that was sheared to
short fragments of similar size to those in ancient samples. In details,
DNA was extracted from the blood of a healthy human donor using the
Gentra Puregene Blood Kit (Qiagen). One microgram of DNA was sheared for
2 h using the Covaris S2 ultrasonicator (shearing parameters: intensity
5, cycler/burst 1000, duty cycles 10%) to obtain a fragment size
distribution that mimics that of ancient DNA. A 200-ng aliquot of
sheared DNA was then used as input for silica-based DNA extraction [3]. A single-stranded library [33]
was prepared from 2.5 μl of the resulting DNA extract (5% of the
extract). The library was amplified using Accuprime Pfx DNA polymerase
(Thermo Fisher Scientific) [34] and a pair of indexing primers containing a sample-specific combination of 7-bp indices [35].
The indexed library was sequenced on 6 lanes of a HiSeq 2000 (Illumina)
in 2 × 76 bp paired-end configuration with two index reads [35].
Sequences without perfect matches to the expected index combination
were discarded. Subsequent processing was carried out identically to the
Mezmaiskaya 1 and Sima de los Huesos data.
For
our positive control—i.e. a sample with solely spurious alignments—we
used 3860 bacteria genomes from the European Nucleotide Archive listed
here http://www.ebi.ac.uk/genomes/bacteria.details.txt.
The genomes were then fragmented from 20 to 40 bp with approximatively
the same number of sequences at each sequence length. This resulted in a
total of ~ 3.03 billion unique sequences (Additional file 1:
Table S1). Given that bacteria might not be the only organisms
representing the environmental contamination, we also used two
eukaryotic genomes. These are a fungus (Saccharomyces cerevisiae,
“sacCer3” S288c strain assembly from GCA_000146055.2) and a protist
(Albugo laibachii, NCBI:txid653948), which resulted in a total of ~ 9.66
and ~ 32.04 million sequences, respectively (Additional file 1: Tables S2 and S3). Ambiguous bases were replaced with one randomly chosen representative base.
All sequence data were mapped to the modified human reference genome using bwa [36] with options “-n 0.01 –o 2 –l 16500” matching those used for the ancient samples [6, 14]. Sequences were merged when they appeared to originate from a PCR duplicate by means of bam-rmdup (https://bitbucket.org/ustenzel/biohazard-tools). Paired-end sequences and sequences shorter than 20 bp were disregarded.
Length-dependent mappability tracks
We used the software GEM [37]
to generate maps of unique mappability of different lengths for the
human reference genome (GCRh37/hg19) including decoy sequences [31].
The program was run for lengths of 20, 23, 26, 29, 32, and 35 bp
allowing for up to one mismatch in alignments. To determine whether a
sequence was mappable, we first chose the largest mappability track that
was not longer than the sequence length. The sequence was deemed
uniquely aligned if it contained a uniquely mappable motif in the
reference within its alignment. All analyses involve this filtering.
Features of spurious alignments
Sequences
that mapped to the modified reference genome and overlap mutated sites
were used to determine characteristics of spurious and true alignments.
Alignments were classified as true if they showed the human reference
base and as spurious if they showed the mutated variant or any other
allele than the human reference. For both spurious and true alignments,
we calculated:
1)
The proportion of
mismatches, i.e., the number of observed mismatches relative to the
modified reference genome divided by sequence length. For sequences that
did not match the modified reference’s allele, we subtracted one
mismatch to compensate for the mismatch caused by the artificially
mutated site. This correction was applied to true and spurious
alignments, alike.
2)
The number of insertion and deletions (indels), extracted from the CIGAR field in the bam/sam format files.
3)
The patterns of nucleotide substitutions, determined by comparing the sequences to the unmodified hg19 reference.
To
minimize the impact of cytosine deamination, we make use of the
preserved strand orientation of sequences prepared with the
single-stranded library protocol [33]
and disregarded alignments in the forward orientation if either the
mutated or original human reference state was C, or alignments in
reverse orientation if the mutated or original human reference state was
G. Due to this filter, 37% of mutated sites (C-to-G or G-to-C) are
disregarded.
Quantifying the fraction of spurious alignments
To calculate the proportion of spurious alignments, we make use of the number of alignments classified as truly related (NT) and the number of alignments classified as spurious (N¬T)
as described in the previous section. A small fraction of spurious
alignments is expected to show the human reference base by chance. To
correct for this, we assume that all spurious alignments contain the
maximum number of mismatches. The maximum proportion of mismatches for a
sequence is M = m/l where m denotes the maximum number of mismatches allowed in a sequence of length l.
Only a third of the exchanges at any given position will match the
original reference base, so that the probability for a spuriously
aligning sequence to show the reference base is at most M × 1/3. We then conservatively correct the NT and N¬T counts to compensate for spurious misclassified alignments by calculating:
𝑁′T=𝑁T−𝑁¬T𝑀3−𝑀
𝑁′¬T=𝑁¬T1−𝑀3
With these corrected counts, we calculate the spurious alignment proportion as:
𝑁′¬T𝑁′¬T+𝑁′.T
Lineage assignment
Informative
sites were determined by sampling one random allele from each of the
genotypes of a modern human (Mbuti, HGDP0456 in [20]), the Altai Neandertal and the Denisovan genomes after applying the minimum set of filters described in [20]. For the Altai Neandertal and Denisovan genomes [21], we used the most recent genotype calls by means of snpAD [38] instead of those of the first publications [6, 20].
To call the ancestral state at each site, we used whole genome
alignments of five primates (pantro4, bonobo, gorgor3, ponabe2, and
rhemac2) to the human reference, and required that at least four of them
agree. Derived sites were assigned to the following four lineages:
Modern Human, Neandertal, Denisovan, and Neandertal-Denisovan.
For
each dataset, we iterated overall sequences, aligned to the unmodified
hg19 reference, and calculated the percentage of derived alleles of each
class that are shared. All T within the last three terminal positions
of sequences were disregarded to minimize the impact of C-to-T changes
due to deamination.
We
are thankful to Michael Dannemann, Janet Kelso, Fabrizio Mafessoni,
Svante Pääbo, Frédéric Romagné, Udo Stenzel, and the Neandertal and
Bioinformatics groups of the Evolutionary Genetics department for the
helpful discussions and suggestions during the development of the
project. We are also grateful to Marie Gansauge and Birgit Nickel for
the extraction and library preparation of the modern human sample. We
are also grateful to three anonymous reviewers for their helpful
comments.
Funding
This study was supported by
the Max Planck Society and funded by the Max Planck Foundation grant
“No. 31-12LMP Pääbo” and by the European Research Council grant No.
694707 to Svante Pääbo.
Availability of data and materials
All data generated or
analyzed during this study are included in this published article and
its supplementary information files. The description of the pipeline,
the software and scripts, and the modified human reference genome are
available in https://figshare.com/projects/SpAl/39137 and can also be found under http://bioinf.eva.mpg.de/SpAl/.
Authors’ contributions
MM
and KP designed the study. All authors analyzed the data, interpreted
the results, and wrote the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to participate
The sequencing of human DNA
as a control for ancient DNA was approved by the Ethik-Komission of the
Medical Faculty of the University of Leipzig (sign: 364-14-17112014).
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open AccessThis article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/),
which permits unrestricted use, distribution, and reproduction in any
medium, provided you give appropriate credit to the original author(s)
and the source, provide a link to the Creative Commons license, and
indicate if changes were made. The Creative Commons Public Domain
Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
Supplementary Information:
Quantifying and reducing spurious alignments for the analysis of
ultra-short ancient DNA sequences
Cesare de Filippo
1
,
∗
, Matthias Meyer
1
, Kay Pr ̈ufer
1
,
∗
1 Max Planck Institute for Evolutionary Anthropology, Leipzig, Germany
∗
Corresponding e-mail: cesare
filippo(at)eva.mpg.de, pruefer(at)eva.mpg.de
1
Table S1. Summary of bacterial sequences.
Length
All unique seq.
Mapped (%)
Classified spurious (%)
Spurious expected (%)
¶
20
143,050,589
91.362
98.10
96.67
21
143,305,294
76.659
98.18
96.83
22
143,476,993
96.892
97.59
95.45
23
143,633,334
89.772
97.63
95.65
24
143,767,397
73.932
97.50
95.83
25
143,881,911
52.541
97.35
96.00
26
143,993,048
32.013
97.29
96.15
27
144,098,076
16.993
97.28
96.30
28
144,191,391
8.126
97.26
96.43
29
144,289,373
3.598
97.23
96.55
30
144,376,099
1.515
97.16
96.67
31
144,458,818
0.622
97.09
96.77
32
144,535,683
0.255
96.90
96.88
33
144,614,643
0.107
96.76
96.97
34
144,682,942
0.047
96.38
97.06
35
144,754,552
0.023
95.57
97.14
36
144,815,313
0.012
93.78
97.22
37
144,882,597
0.007
92.16
97.30
38
144,947,978
0.005
87.05
97.37
39
145,005,407
0.003
82.10
97.44
40
145,061,336
0.003
70.83
97.50
total
3,029,822,774
25.798
97.57
96.06
¶
The expected proportion of spurious alignment assuming that all sequences have the maximum number of
mismatches was calculated as 1-(
M
/3) where
M = m/l
(see Materials and Methods), that is the maximum
proportion of mismatches allowed in the alignment (
m
) at a given length (
l
). This is divided by 3 because three are
all alternative non-reference alleles one of which is the hg19 reference. Using the ‘ancient’ parameters for alignment,
2 and 3 maximum mismatches are allowed for sequences of length 20-21 bp and 22-41 bp, respectively.
Allentoft
ME, Collins M, Harker D, Haile J, Oskam CL, Hale ML, et al. The
half-life of DNA in bone: measuring decay kinetics in 158 dated fossils.
Proc Biol Sci. 2012;279:4724–33.View ArticlePubMedPubMed CentralGoogle Scholar
Dabney
J, Knapp M, Glocke I, Gansauge M-T, Weihmann A, Nickel B, et al.
Complete mitochondrial genome sequence of a Middle Pleistocene cave bear
reconstructed from ultrashort DNA fragments. Proc Natl Acad Sci U A S.
2013;110:15758–63.View ArticleGoogle Scholar
Allentoft
ME, Sikora M, Sjögren K-G, Rasmussen S, Rasmussen M, Stenderup J, et
al. Population genomics of Bronze Age Eurasia. Nature. 2015;522:167–72.View ArticleGoogle Scholar
Meyer
M, Kircher M, Gansauge M-T, Li H, Racimo F, Mallick S, et al. A
high-coverage genome sequence from an archaic Denisovan individual.
Science. 2012;338:222–6.View ArticlePubMedPubMed CentralGoogle Scholar
Meyer
M, Fu Q, Aximu-Petri A, Glocke I, Nickel B, Arsuaga J-L, et al. A
mitochondrial genome sequence of a hominin from Sima de los Huesos.
Nature. 2014;505:403–6.View ArticleGoogle Scholar
Meyer
M, Arsuaga J-L, de Filippo C, Nagel S, Aximu-Petri A, Nickel B, et al.
Nuclear DNA sequences from the Middle Pleistocene Sima de los Huesos
hominins. Nature. 2016;531:504.View ArticleGoogle Scholar
Noonan
JP, Hofreiter M, Smith D, Priest JR, Rohland N, Rabeder G, et al.
Genomic sequencing of Pleistocene cave bears. Science. 2005;309:597–9.View ArticleGoogle Scholar
Green
RE, Krause J, Ptak SE, Briggs AW, Ronan MT, Simons JF, et al. Analysis
of one million base pairs of Neanderthal DNA. Nature. 2006;444:330–6.View ArticleGoogle Scholar
Poinar
HN, Schwarz C, Qi J, Shapiro B, Macphee RDE, Buigues B, et al.
Metagenomics to paleogenomics: large-scale sequencing of mammoth DNA.
Science. 2006;311:392–4.View ArticleGoogle Scholar
Der
Sarkissian C, Allentoft ME, Ávila-Arcos MC, Barnett R, Campos PF,
Cappellini E, et al. Ancient genomics. Philos Trans R Soc Lond Ser B
Biol Sci. 2015;370:20130387.View ArticleGoogle Scholar
Prüfer
K, Stenzel U, Hofreiter M, Pääbo S, Kelso J, Green RE. Computational
challenges in the analysis of ancient DNA. Genome Biol. 2010;11:R47.View ArticlePubMedPubMed CentralGoogle Scholar
Green
RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, et al. A
draft sequence of the Neandertal genome. Science. 2010;328:710–22.View ArticlePubMedPubMed CentralGoogle Scholar
Schubert
M, Ginolhac A, Lindgreen S, Thompson JF, Al-Rasheid KAS, Willerslev E,
et al. Improving ancient DNA read mapping against modern reference
genomes. BMC Genomics. 2012;13:178.View ArticlePubMedPubMed CentralGoogle Scholar
Bennett
EA, Massilani D, Lizzo G, Daligault J, Geigl E-M, Grange T. Library
construction for ancient genomics: single strand or double strand?
BioTechniques. 2014;56:289–300.View ArticleGoogle Scholar
Renaud G, Hanghøj K, Willerslev E, Orlando L. gargammel: a sequence simulator for ancient DNA. Bioinformatics. 2017;33:577–9.Google Scholar
Prüfer
K, Racimo F, Patterson N, Jay F, Sankararaman S, Sawyer S, et al. The
complete genome sequence of a Neanderthal from the Altai Mountains.
Nature. 2014;505:43–9.View ArticleGoogle Scholar
Prüfer
K, de Filippo C, Grote S, Mafessoni F, Korlević P, Hajdinjak M, et al. A
high-coverage Neandertal genome from Vindija Cave in Croatia. Science.
2017;358:655–8.View ArticlePubMedPubMed CentralGoogle Scholar
Briggs
AW, Stenzel U, Johnson PLF, Green RE, Kelso J, Prüfer K, et al.
Patterns of damage in genomic DNA sequences from a Neandertal. Proc Natl
Acad Sci U S A. 2007;104:14616–21.View ArticlePubMedPubMed CentralGoogle Scholar
Green
RE, Briggs AW, Krause J, Prüfer K, Burbano HA, Siebauer M, et al. The
Neandertal genome and ancient DNA authenticity. EMBO J.
2009;28:2494–502.View ArticlePubMedPubMed CentralGoogle Scholar
Reich
D, Green RE, Kircher M, Krause J, Patterson N, Durand EY, et al.
Genetic history of an archaic hominin group from Denisova cave in
Siberia. Nature. 2010;468:1053–60.View ArticlePubMedPubMed CentralGoogle Scholar
Reich
D, Patterson N, Kircher M, Delfin F, Nandineni MR, Pugach I, et al.
Denisova admixture and the first modern human dispersals into Southeast
Asia and Oceania. Am J Hum Genet. 2011;89:516–28.View ArticlePubMedPubMed CentralGoogle Scholar
Benjamini
Y, Hochberg Y. Controlling the false discovery rate: a practical and
powerful approach to multiple testing. J R Stat Soc Ser B Stat Methodol.
1995;57:289–300.Google Scholar
Fu
Q, Meyer M, Gao X, Stenzel U, Burbano HA, Kelso J, et al. DNA analysis
of an early modern human from Tianyuan Cave, China. Proc Natl Acad Sci U
S A. 2013;110:2223–7.View ArticlePubMedPubMed CentralGoogle Scholar
Damgaard
PB, Margaryan A, Schroeder H, Orlando L, Willerslev E, Allentoft ME.
Improving access to endogenous DNA in ancient bones and teeth. Sci Rep.
2015;5:11184.View ArticlePubMedPubMed CentralGoogle Scholar
Korlević
P, Gerber T, Gansauge M-T, Hajdinjak M, Nagel S, Aximu-Petri A, et al.
Reducing microbial and human contamination in DNA extractions from
ancient bones and teeth. BioTechniques. 2015;59:87–93.View ArticleGoogle Scholar
1000
Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP,
Kang HM, et al. A global reference for human genetic variation. Nature.
2015;526:68–74.View ArticleGoogle Scholar
Gansauge
M-T, Meyer M. Single-stranded DNA library preparation for the
sequencing of ancient or damaged DNA. Nat Protoc. 2013;8:737–48.View ArticleGoogle Scholar
Dabney
J, Meyer M. Length and GC-biases during sequencing library
amplification: a comparison of various polymerase-buffer systems with
ancient and modern DNA sequencing libraries. BioTechniques.
2012;52:87–94.View ArticleGoogle Scholar
Kircher
M, Sawyer S, Meyer M. Double indexing overcomes inaccuracies in
multiplex sequencing on the Illumina platform. Nucleic Acids Res.
2012;40:e3.View ArticleGoogle Scholar
Marco-Sola
S, Sammeth M, Guigó R, Ribeca P. The GEM mapper: fast, accurate and
versatile alignment by filtration. Nat Methods. 2012;9:1185–8.View ArticlePubMedGoogle Scholar
Malaspinas
A-S, Lao O, Schroeder H, Rasmussen M, Raghavan M, Moltke I, et al. Two
ancient human genomes reveal Polynesian ancestry among the indigenous
Botocudos of Brazil. Curr Biol. 2014;24:1035–7.View ArticleGoogle Scholar
Raghavan
M, Skoglund P, Graf KE, Metspalu M, Albrechtsen A, Moltke I, et al.
Upper Palaeolithic Siberian genome reveals dual ancestry of Native
Americans. Nature. 2014;505:87–91.View ArticleGoogle Scholar
Martiniano
R, Caffell A, Holst M, Hunter-Mann K, Montgomery J, Müldner G, et al.
Genomic signals of migration and continuity in Britain before the
Anglo-Saxons. Nat Commun. 2016;7:10326.View ArticlePubMedPubMed CentralGoogle Scholar
Raghavan
M, Steinrücken M, Harris K, Schiffels S, Rasmussen S, DeGiorgio M, et
al. Genomic evidence for the Pleistocene and recent population history
of Native Americans. Science. 2015;349:aab3884.View ArticlePubMedPubMed CentralGoogle Scholar
Rasmussen
M, Guo X, Wang Y, Lohmueller KE, Rasmussen S, Albrechtsen A, et al. An
aboriginal Australian genome reveals separate human dispersals into
Asia. Science. 2011;334:94–8.View ArticlePubMedPubMed CentralGoogle Scholar
Gamba
C, Jones ER, Teasdale MD, McLaughlin RL, Gonzalez-Fortes G, Mattiangeli
V, et al. Genome flux and stasis in a five millennium transect of
European prehistory. Nat Commun. 2014;5:5257.View ArticlePubMedPubMed CentralGoogle Scholar
Olalde
I, Allentoft ME, Sánchez-Quinto F, Santpere G, Chiang CWK, DeGiorgio M,
et al. Derived immune and ancestral pigmentation alleles in a
7,000-year-old Mesolithic European. Nature. 2014;507:225–8.View ArticlePubMedPubMed CentralGoogle Scholar
Gallego
Llorente M, Jones ER, Eriksson A, Siska V, Arthur KW, Arthur JW, et al.
Ancient Ethiopian genome reveals extensive Eurasian admixture
throughout the African continent. Science. 2015;350:820–2.View ArticleGoogle Scholar
Jones
ER, Gonzalez-Fortes G, Connell S, Siska V, Eriksson A, Martiniano R, et
al. Upper Palaeolithic genomes reveal deep roots of modern Eurasians.
Nat Commun. 2015;6:8912.View ArticlePubMedPubMed CentralGoogle Scholar
Olalde
I, Schroeder H, Sandoval-Velasco M, Vinner L, Lobón I, Ramirez O, et
al. A common genetic origin for early farmers from Mediterranean Cardial
and Central European LBK cultures iterranean Cardial and Central
European LBK cultures. Mol Biol Evol. 2015;32:3132–42.PubMedPubMed CentralGoogle Scholar
Rasmussen
M, Sikora M, Albrechtsen A, Korneliussen TS, Moreno-Mayar JV, Poznik
GD, et al. The ancestry and affiliations of Kennewick Man. Nature.
2015;523:455–8.PubMedPubMed CentralGoogle Scholar
Hofmanová
Z, Kreutzer S, Hellenthal G, Sell C, Diekmann Y, Díez-Del-Molino D, et
al. Early farmers from across Europe directly descended from Neolithic
Aegeans. Proc Natl Acad Sci U S A. 2016;113:6886–91.View ArticlePubMedPubMed CentralGoogle Scholar
Cassidy
LM, Martiniano R, Murphy EM, Teasdale MD, Mallory J, Hartwell B, et al.
Neolithic and Bronze Age migration to Ireland and establishment of the
insular Atlantic genome. Proc Natl Acad Sci U S A. 2016;113:368–73.View ArticleGoogle Scholar
Fu
Q, Li H, Moorjani P, Jay F, Slepchenko SM, Bondarev AA, et al. Genome
sequence of a 45,000-year-old modern human from western Siberia. Nature.
2014;514:445–9.View ArticlePubMedPubMed CentralGoogle Scholar
Lazaridis
I, Patterson N, Mittnik A, Renaud G, Mallick S, Kirsanow K, et al.
Ancient human genomes suggest three ancestral populations for
present-day Europeans. Nature. 2014;513:409–13.View ArticlePubMedPubMed CentralGoogle Scholar
Skoglund
P, Malmström H, Omrak A, Raghavan M, Valdiosera C, Günther T, et al.
Genomic diversity and admixture differs for Stone-Age Scandinavian
foragers and farmers. Science. 2014;344:747–50.View ArticleGoogle Scholar
Fu
Q, Hajdinjak M, Moldovan OT, Constantin S, Mallick S, Skoglund P, et
al. An early modern human from Romania with a recent Neanderthal
ancestor. Nature. 2015;524:216–9.View ArticlePubMedPubMed CentralGoogle Scholar
Günther
T, Valdiosera C, Malmström H, Ureña I, Rodriguez-Varela R,
Sverrisdóttir ÓO, et al. Ancient genomes link early farmers from
Atapuerca in Spain to modern-day Basques. Proc Natl Acad Sci U A.
2015;112:11917–22.View ArticleGoogle Scholar
Schlebusch
CM, Malmström H, Günther T, Sjödin P, Coutinho A, Edlund H, et al.
Southern African ancient genomes estimate modern human divergence to
350,000 to 260,000 years ago. Science. 2017;358:652–5.View ArticlePubMedPubMed CentralGoogle Scholar
Skoglund
P, Thompson JC, Prendergast ME, Mittnik A, Sirak K, Hajdinjak M, et al.
Reconstructing Prehistoric African Population Structure. Cell.
2017;171:59–71 e21.View ArticlePubMedPubMed CentralGoogle Scholar
Hajdinjak
M, Fu Q, Hübner A, Petr M, Mafessoni F, Grote S, et al. Reconstructing
the genetic history of late Neanderthals. Nature. 2018;555:652–6.View ArticleGoogle Scholar
Rasmussen
M, Li Y, Lindgreen S, Pedersen JS, Albrechtsen A, Moltke I, et al.
Ancient human genome sequence of an extinct Palaeo-Eskimo. Nature.
2010;463:757–62.View ArticlePubMedPubMed CentralGoogle Scholar
Keller
A, Graefen A, Ball M, Matzas M, Boisguerin V, Maixner F, et al. New
insights into the Tyrolean Iceman’s origin and phenotype as inferred by
whole-genome sequencing. Nat Commun. 2012;3:698.View ArticleGoogle Scholar
Rasmussen
M, Anzick SL, Waters MR, Skoglund P, DeGiorgio M, Stafford TW Jr, et
al. The genome of a Late Pleistocene human from a Clovis burial site in
western Montana. Nature. 2014;506:225–9.View ArticlePubMedPubMed CentralGoogle Scholar
Seguin-Orlando
A, Korneliussen TS, Sikora M, Malaspinas A-S, Manica A, Moltke I, et
al. Paleogenomics. Genomic structure in Europeans dating back at least
36,200 years. Science. 2014;346:1113–8.View ArticleGoogle Scholar
Schiffels
S, Haak W, Paajanen P, Llamas B, Popescu E, Loe L, et al. Iron Age and
Anglo-Saxon genomes from East England reveal British migration history.
Nat Commun. 2016;7:10408.View ArticlePubMedPubMed CentralGoogle Scholar
Sikora
M, Seguin-Orlando A, Sousa VC, Albrechtsen A, Korneliussen T, Ko A, et
al. Ancient genomes show social and reproductive behavior of early upper
Paleolithic foragers. Science. 2017;358:659–62.
Nenhum comentário:
Postar um comentário
Observação: somente um membro deste blog pode postar um comentário.
Nenhum comentário:
Postar um comentário
Observação: somente um membro deste blog pode postar um comentário.