Os genomas de cães europeus antigos revelam continuidade desde o início do neolítico
Nature Communications, volume 8 , número do artigo: 16082 ( 2017 )
Abstrato
A Europa desempenhou um papel importante na evolução dos cães,
abrigando os restos paleolíticos mais antigos e incontestados e tendo
sido o centro da criação moderna de raças de cães.
Aqui, sequenciamos os genomas de um cão neolítico precoce e final da
Alemanha, incluindo uma amostra associada a uma comunidade agrícola europeia antiga.
Ambos os cães demonstram continuidade um com o outro e compartilham
predominantemente ancestralidade com os cães europeus modernos,
contradizendo uma substituição populacional neolítica tardia sugerida
anteriormente. Não encontramos evidências genéticas para apoiar a hipótese recente que propõe origens duplas da domesticação de cães.
Ao calibrar a taxa de mutação usando nosso cão mais velho, reduzimos o
tempo da domesticação do cão para 20.000 a 40.000 anos atrás. Curiosamente, não observamos a expansão extrema do número de cópias do gene AMY2B,
característica de cães modernos, que havia sido proposta anteriormente
como uma adaptação a uma dieta rica em amido, impulsionada pela ampla
adoção da agricultura no Neolítico.
Introdução
A Europa tem sido uma região criticamente importante na história e
evolução dos cães, com a maioria das raças modernas compartilhando
predominantemente ascendência europeia 1 . Além disso, os restos mais antigos que podem ser inequivocamente atribuídos a cães domésticos ( Canis lupus familiaris
) são encontrados neste continente, incluindo uma mandíbula do osso
paleolítico superior de 14.700 anos do local de Bonn – Oberkassel na
Alemanha 2 (espécimes mais antigos da Sibéria e o Oriente Próximo proposto permanece altamente controverso 3 , 4 ). Enquanto o antigo DNA mitocondrial (mtDNA) sugere um centro europeu de domesticação de cães 5 , análises de dados mitocondriais e genômicos de cães modernos sugeriram o Leste da Ásia 6 , 7 , o Oriente Médio 8 e a Ásia Central 9 .
O período neolítico na Europa Central varia de 7.500 a 4.000 PA e pode ser subdividido com base em características específicas da cultura humana 10 ( Tabela Suplementar 1 ). Vários estudos encontraram evidências de uma rotatividade pré-histórica de linhagens de mtDNA canídeos em algum momento entre o Neolítico tardio e hoje, com o haplogrupo C, que aparece em quase todos os cães neolíticos, mas em menos de 10% dos cães modernos, sendo substituído pelo haplogrupo A na maioria dos países. Europa 5 , 11 , 12 . Ao analisar dados genômicos de cães modernos e um cão irlandês neolítico tardio ( ∼ 5.000 anos) de Newgrange (doravante denominado NGD), Frantz et al . 12 argumentam que essa rotatividade matrilinear foi consequência de uma grande substituição populacional durante o Neolítico. No entanto, o NGD compartilha principalmente a ancestralidade com os cães europeus modernos, o que implica que a substituição populacional proposta já havia ocorrido amplamente antes desse indivíduo viver. Frantz et al . 12 também estimam uma divergência relativamente recente entre cães leste-oeste (14.000 a 6.000 anos atrás), que, colocada no contexto dos dados arqueológicos existentes, explica com uma origem dupla da domesticação de cães.
A caracterização de amostras anteriores no Neolítico e na Europa continental é necessária para examinar se e até que ponto ocorreu uma substituição demográfica em larga escala durante esse período. Isso seria evidenciado por uma ancestralidade distinta, ausente nos genomas de cães modernos, que foi mais proeminente em cães do início do Neolítico, em oposição à continuidade genômica do Neolítico Inicial até hoje. Portanto, apresentamos a análise de genomas inteiros de 9 × coberturas de duas amostras de cães da Alemanha datando do Neolítico Precoce e Final ( ∼ 7.000 anos e ∼ 4.700 anos, respectivamente). Observamos a continuidade genética ao longo desta época e até o presente, com nossos cães antigos compartilhando ascendência substancial com os cães europeus modernos. Não encontramos evidências de uma grande substituição populacional; em vez disso, nossos resultados são consistentes com um cenário em que os cães europeus modernos emergiram de uma população neolítica estruturada. Além disso, detectamos um componente de ancestralidade adicional na amostra do Neolítico Final, consistente com a mistura de uma população de cães localizados mais a leste que pode ter migrado concomitantemente com pessoas da estepe associadas a culturas da Idade do Neolítico Tardio e do Início do Bronze, como Yamnaya e Corded Ware cultura 13 . Também mostramos que a maioria dos haplótipos autossômicos associados à domesticação já foi estabelecida em nossos cães neolíticos, mas provavelmente a adaptação a uma dieta rica em amido ocorreu mais tarde. Finalmente, obtemos estimativas de divergência entre cães orientais e ocidentais de 17.000 a 24.000 anos atrás, consistentes com uma única origem geográfica para domesticação, cujo período de tempo reduzimos para entre 20.000 e 40.000 anos atrás.
O período neolítico na Europa Central varia de 7.500 a 4.000 PA e pode ser subdividido com base em características específicas da cultura humana 10 ( Tabela Suplementar 1 ). Vários estudos encontraram evidências de uma rotatividade pré-histórica de linhagens de mtDNA canídeos em algum momento entre o Neolítico tardio e hoje, com o haplogrupo C, que aparece em quase todos os cães neolíticos, mas em menos de 10% dos cães modernos, sendo substituído pelo haplogrupo A na maioria dos países. Europa 5 , 11 , 12 . Ao analisar dados genômicos de cães modernos e um cão irlandês neolítico tardio ( ∼ 5.000 anos) de Newgrange (doravante denominado NGD), Frantz et al . 12 argumentam que essa rotatividade matrilinear foi consequência de uma grande substituição populacional durante o Neolítico. No entanto, o NGD compartilha principalmente a ancestralidade com os cães europeus modernos, o que implica que a substituição populacional proposta já havia ocorrido amplamente antes desse indivíduo viver. Frantz et al . 12 também estimam uma divergência relativamente recente entre cães leste-oeste (14.000 a 6.000 anos atrás), que, colocada no contexto dos dados arqueológicos existentes, explica com uma origem dupla da domesticação de cães.
A caracterização de amostras anteriores no Neolítico e na Europa continental é necessária para examinar se e até que ponto ocorreu uma substituição demográfica em larga escala durante esse período. Isso seria evidenciado por uma ancestralidade distinta, ausente nos genomas de cães modernos, que foi mais proeminente em cães do início do Neolítico, em oposição à continuidade genômica do Neolítico Inicial até hoje. Portanto, apresentamos a análise de genomas inteiros de 9 × coberturas de duas amostras de cães da Alemanha datando do Neolítico Precoce e Final ( ∼ 7.000 anos e ∼ 4.700 anos, respectivamente). Observamos a continuidade genética ao longo desta época e até o presente, com nossos cães antigos compartilhando ascendência substancial com os cães europeus modernos. Não encontramos evidências de uma grande substituição populacional; em vez disso, nossos resultados são consistentes com um cenário em que os cães europeus modernos emergiram de uma população neolítica estruturada. Além disso, detectamos um componente de ancestralidade adicional na amostra do Neolítico Final, consistente com a mistura de uma população de cães localizados mais a leste que pode ter migrado concomitantemente com pessoas da estepe associadas a culturas da Idade do Neolítico Tardio e do Início do Bronze, como Yamnaya e Corded Ware cultura 13 . Também mostramos que a maioria dos haplótipos autossômicos associados à domesticação já foi estabelecida em nossos cães neolíticos, mas provavelmente a adaptação a uma dieta rica em amido ocorreu mais tarde. Finalmente, obtemos estimativas de divergência entre cães orientais e ocidentais de 17.000 a 24.000 anos atrás, consistentes com uma única origem geográfica para domesticação, cujo período de tempo reduzimos para entre 20.000 e 40.000 anos atrás.
Resultados
Amostras arqueológicas e sequenciamento antigo de DNA
O espécime mais antigo, ao qual nos referimos a seguir como HXH, foi encontrado no local neolítico inicial de Herxheim e tem data de 5.223-5.040 cal. AEC ( ± 7.000 anos) ( Fig. 1 complementar ). O espécime mais jovem, a que nos referimos a seguir como CTC, foi encontrado na caverna da árvore das cerejeiras e é de 2.900 a 2.632 cal. AEC ( ∼ 4.700 anos), que corresponde ao período Neolítico Final na Europa Central 14 ( Fig. 2 e Notas Suplementares 1–3 ).Geramos dados da sequência do genoma inteiro para os dois cães antigos e mapeamos mais de 67% das leituras para o genoma de referência do cão (CanFam3.1), confirmando o alto conteúdo de DNA canino endógeno das duas amostras ( Tabela Suplementar 2 e Nota Suplementar 4 ). A análise do MapDamage 15 demonstrou que ambas as amostras possuem características de dano típicas do DNA antigo 16 ( Fig. 3 complementar ). A cobertura média final para ambas as amostras foi de ± 9 ×, enquanto a cobertura no cromossomo X e Y foi de ± 5 ×, indicando que ambos são do sexo masculino. Também reprocessamos os dados de NGD 12 usando o mesmo pipeline que para CTC e HXH. Para chamar variantes, usamos um identificador de genótipo personalizado implementado em Python (consulte a Nota Suplementar 5 ) que é responsável pelos padrões de danos ao DNA 17 . Descobrimos que nossa abordagem eliminou muitos falsos positivos que provavelmente são causados por danos post-mortem ( Figura 4 ).
Conjuntos de dados de referência canid modernos
Analisamos esses cães neolíticos no contexto de uma coleção abrangente de 5.649 canídeos, incluindo cães de raças, cães de vila e lobos que haviam sido previamente genotipados em 128.743 polimorfismos de nucleotídeo único (SNPs) 9 , 18 ( Tabela complementar 3 ), bem como 99 genomas inteiros canídeos sequenciados com cobertura média a alta (6-45 ×) ( Tabela Suplementar 4 ). Para explicar os vieses na chamada de variantes que podem ocorrer como resultado dessa cobertura variável, verificamos locais variáveis em um grupo externo (de modo que se sabe que mutações ocorreram na raiz de todas as populações analisadas). Exploramos diferentes esquemas de verificação para os dados de todo o genoma ( Nota Complementar 6 ) e optamos por usar um conjunto de chamadas que inclui sites variáveis nos lobos do Novo Mundo (observamos, porém, que nossos principais resultados são robustos a alterações no esquema de verificação) . Esse conjunto de chamadas contém 1.815.911 variantes que provavelmente são privadas dos lobos do Novo Mundo ou surgiram na população ancestral do lobo cinza ( Canis lupus ) e, portanto, são as menos tendenciosas em relação à sua constatação em lobos e cães do Velho Mundo.análise mtDNA
Examinamos a relação filogenética de todos os genomas mitocondriais de HXH e CTC com um painel abrangente de cães modernos em quatro grandes clados (A-D), lobos e coiotes modernos e sequências mitocondriais inteiras semelhantes a cães e cães 5 , 12 . Como outros cães neolíticos europeus, tanto o HXH quanto o CTC pertencem ao haplogrupo C ( Fig. 1a , Fig. Complementar 5 e Nota complementar 7 ) juntamente com o NGD e o cão da caverna Kartstein de 12.500 anos do Paleolítico Superior (também da Alemanha). Observamos que Bonn – Oberkassel também cai no mesmo haplogrupo 5 ( Figs. 6 e 7 Complementares ), embora a análise desta amostra seja complicada pela baixa cobertura da sequência do mtDNA, apontando para algum grau de continuidade matrilinear na Europa por mais de 10.000 anos, variando de o paleolítico tardio a quase todo o neolítico. A inclusão de 24 amostras adicionais de clado C 19 na análise filogenética revela a divisão C1 e C2 esperada (suporte a 100%) e que HXH, CTC, NGD e o cão da caverna de Kartstein compartilham uma linhagem comum com os cães C1 ( Fig. 8 ). . Esta topologia sugere que esses cães europeus antigos pertencem a um sub-haplogrupo mais antigo, que é irmã do progenitor dos sub-haplogrupos C1b e C1a e possivelmente ausente nas populações modernas de cães.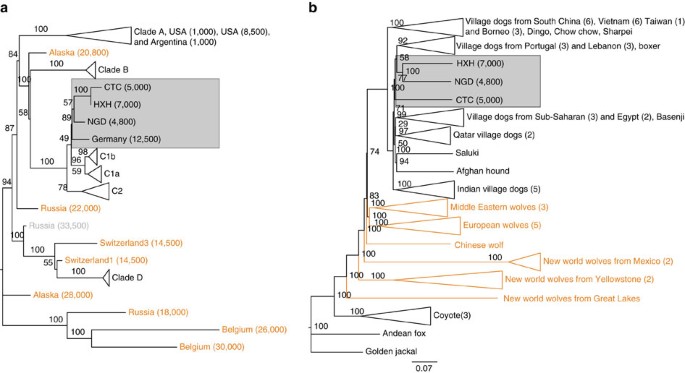
( a ) Filogenia baseada em mtDNA. A idade das amostras é indicada entre parênteses, as amostras de lobo são mostradas em laranja. ( b ) árvore NJ baseada na divergência de sequência em pares dos dados do genoma inteiro.
Agrupamento genômico dos cães neolíticos europeus
Construímos uma árvore de união de vizinhos (NJ) usando o conjunto de dados da sequência do genoma inteiro ( Fig. 1b e Figs. 9 e 10 complementares ) para determinar qual população de cães modernos apresenta a maior semelhança genética com as amostras antigas ( Nota complementar 8 ). Descobrimos que o HXH neolítico precoce e o NGD neolítico tardio agrupavam-se como um clã irmão dos modernos cães da aldeia da Europa, enquanto o CTC era externo a esse clado, mas ainda mais semelhante a ele do que qualquer outra população moderna. Como mostrado anteriormente 12 , os cães e raças do leste asiático são básicos para todos os outros cães.Também realizamos uma análise de componentes principais (PCA) usando a matriz SNP e os dados do genoma inteiro ( Fig. 2a, be Nota Complementar 9 ), com ambos os conjuntos de dados mostrando padrões de estrutura populacional altamente semelhantes, apesar de terem esquemas de apuração muito diferentes ( Espera-se que os dados da matriz SNP sejam direcionados para cães de raças européias). O conjunto maior de dados de referência da matriz SNP mostra que os cães das aldeias se separam principalmente em cinco grupos geográficos distintos: Sudeste Asiático, Índia, Oriente Médio, Europa e África. Os cães de raças se enquadram principalmente na variação dos cães de vilarejos europeus, com exceção das raças basais ou "antigas" 20 . Consistente com a análise de árvores de NJ, todas as três amostras antigas caíram dentro do intervalo da variação moderna de cães. HXH e NGD são as amostras antigas encontradas mais próximas ao maior aglomerado europeu, ambas localizadas adjacentes ao aglomerado de cães das Ilhas do Pacífico que, acredita-se, são derivadas quase completamente dos cães europeus 9 . O CTC está localizado próximo aos cães da aldeia do Afeganistão, uma população conhecida e misturada que também se deduz ter um importante componente de ascendência do tipo europeu, além de possíveis contribuições das populações do sul e do leste da Ásia 9 .
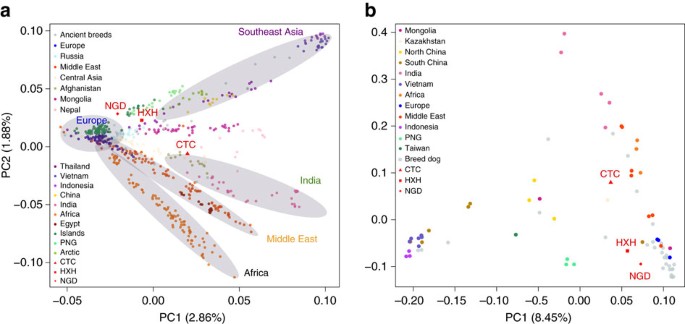
( a ) PCA de cães da vila, com cães de raças e cães antigos projetados no espaço do PC usando dados de matriz SNP. ( b ) PCA de cães da vila, cães de raças e cães antigos, usando dados SNP de todo o genoma apurados nos lobos do Novo Mundo.
Examinamos ainda a relação genética entre cães antigos e modernos, realizando uma análise do grupo f3 21 , 22 nos conjuntos de dados da matriz SNP e da seqüência do genoma inteiro. Utilizamos o chacal de ouro e a raposa andina como grupos externos para a matriz SNP e os conjuntos de dados de todo o genoma, respectivamente. Nossos resultados corroboram os achados das árvores NJ e da PCA e mostram que todas as três amostras européias neolíticas são geneticamente mais semelhantes aos cães europeus modernos ( Fig. 3 , Figs. 13 e 14 complementares e Nota complementar 10 ).
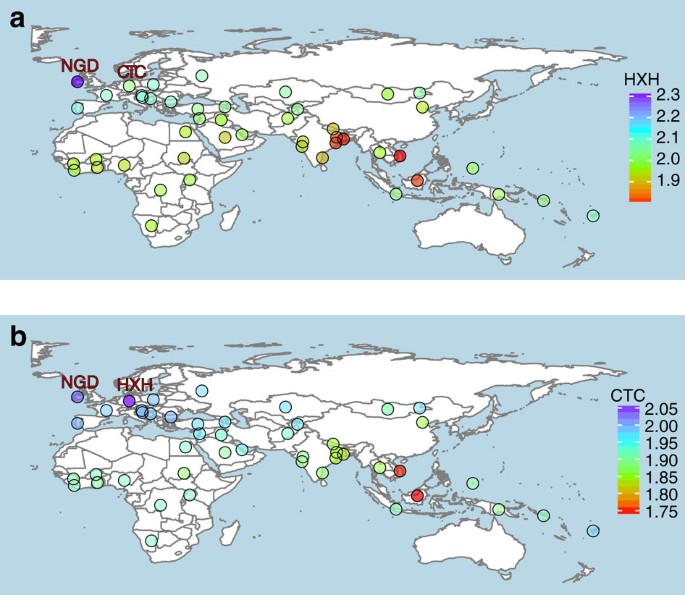
Mapa de calor das estatísticas do grupo f3 do grupo f3 (chacal dourado; amostra antiga; X) com base nos dados do genótipo do arranjo SNP. Valores mais altos de f3 indicam aumento da deriva compartilhada entre as amostras e, portanto, semelhanças genéticas mais altas. ( a ) O HXH mostra maior semelhança com NGD e cães modernos da aldeia européia (valores mais altos de f3 ) e é mais distante dos cães da vila do leste asiático e indiano. ( b ) O CTC compartilha a maior semelhança genética com o HXH, seguido pelo NGD e outros cães europeus.
Além disso, o CTC mostra uma maior semelhança com os cães da aldeia da
Índia (particularmente populações não misturadas no leste) do que o HXH. Mapas estatísticos do grupo externo f3 foram criados usando os pacotes R ggplot2 e mapas usando o conjunto de dados Natural Earth de domínio público.
Evidência de mistura em cães neolíticos
Nossos resultados são consistentes com a continuidade de uma ascendência genética do tipo europeu dos cães modernos durante todo o período neolítico. No entanto, a posição levemente deslocada das amostras antigas do cluster europeu nos PCAs (particularmente no CTC) sugere uma história complexa. Portanto, realizamos análises de cluster não supervisionadas com ADMIXTURE (dados do arranjo SNP; Fig. 15 suplementar ) e NGSadmix (dados de genoma completo; Fig. 4 e Fig. 16 complementar ) ( Nota complementar 9 ) e descobrimos que, diferentemente dos cães de vilarejo europeus contemporâneos, todos os três genomas antigos possuem um componente significativo de ancestralidade presente nos cães modernos do sudeste asiático. Esse componente aparece apenas em níveis muito baixos em uma minoria de cães de vilarejos europeus modernos. Além disso, a CTC abriga um componente adicional que é encontrado predominantemente em vilarejos indianos modernos, bem como em cães da Ásia Central (afegã, mongol e nepalesa) e do Oriente Médio (Arábia Saudita e Catar) (concordante com sua posição na APC), como bem como alguma mistura de lobo.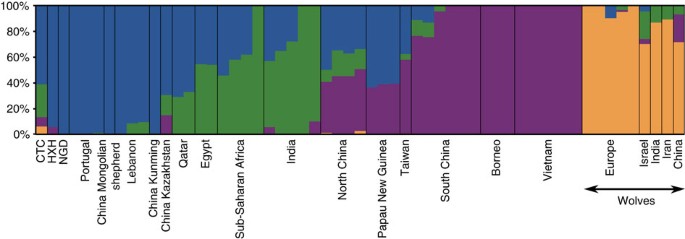
Agrupamento NGSadmix para K = 4 para cães da aldeia, cães antigos e lobos do Velho Mundo, com base nos dados SNP de todo o genoma.
Para separar os padrões de mistura mais complexos observados no CTC, buscamos primeiro entender sua relação com o HXH, uma vez que ambas as amostras são originárias da Alemanha. Nossa análise do grupo f3 revelou que a CTC tinha maior afinidade com o HXH do que com qualquer canídeo moderno ou com NGD ( Fig. 3b , Figs. Suplementares 13b e 14b e Nota Complementar 10 ). Portanto, realizamos uma análise MixMapper em que o HXH foi definido como uma das fontes de mistura para CTC, que identificou uma população ancestral dos modernos cães da vila da Índia ou da Arábia Saudita como a segunda fonte de mistura ( Tabela Suplementar 11 ). Foi encontrado suporte adicional para a continuidade genética entre HXH e CTC usando ADMIXTUREGRAPH quando dois modelos demográficos alternativos foram testados. No primeiro modelo (modelo A), o CTC descende da mesma população que o HXH, seguido de mistura com uma população do tipo indiano, enquanto no modelo B ambas as amostras antigas descendem de linhagens européias divergentes independentemente (e, portanto, não havia continuidade genética entre os dois).
O Modelo A fornece um ajuste muito melhor aos dados ( Fig. 5a e Fig. 20 suplementar ), produzindo apenas dois outliers f4 (sem outliers f2 ou f3 ), um dos quais foi pouco significativo ( Z = 3.013). O Modelo B produziu 74 discrepantes ( Nota Complementar 11 ). Embora exista o risco de sobreajustar o modelo aos dados, a grande diferença entre os dois modelos aponta para a continuidade entre os cães alemães durante o Neolítico, juntamente com o fluxo gênico para a CTC no final desta época, de uma fonte externa portadora do gene genético. componente observado nas populações contemporâneas de cães do Oriente Médio e Central e do Sul da Ásia.
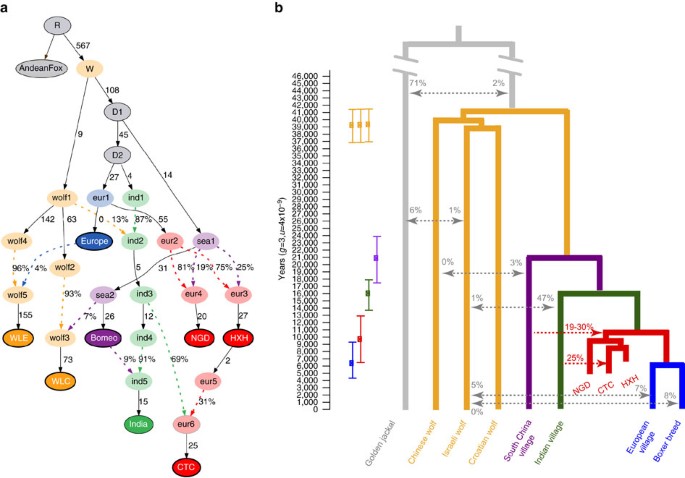
( a
) O melhor modelo se encaixa nos dados de canídeos modernos e antigos
usando ADMIXTUREGRAPH no conjunto de dados de todo o genoma. Este modelo teve quatro outliers estatísticos f4 .
As ramificações são indicadas por linhas pretas sólidas (números
adjacentes indicam valores de desvio estimados em unidades de distância f2
, partes por mil), enquanto a mistura é indicada por linhas tracejadas
coloridas (números adjacentes indicam proporções ancestrais). Os indivíduos / populações amostrados são indicados por círculos sólidos com contorno em negrito. Lobos são rotulados como 'lobo' e cães são rotulados de acordo com sua origem continental. ( b ) Tempos de divergência de cães e lobos contemporâneos inferidos usando G-PhoCS. As estimativas médias são indicadas por quadrados com faixas correspondentes a intervalos credíveis bayesianos de 95%.
As faixas de migração são mostradas em cinza com o valor associado que
representa as taxas de migração total inferidas (a probabilidade de uma
linhagem na população alvo migrar para a população de origem). O tempo de divergência para HXH e NGD e cães europeus modernos é inferido usando uma abordagem numérica.
A proporção de ascendência indiana de cães de aldeia na CTC é inferida
por NGSadmix e a proporção de ascendência de cães de vila do sul da
China em HXH e NGD é inferida pelo teste de f4- rácio, mostrado em vermelho.
O padrão complexo de mistura encontrado na CTC é semelhante ao observado em muitas populações modernas de cães na Ásia Central (como o Afeganistão) e no Oriente Médio, como mostrado em nossas análises de agrupamento não supervisionadas ( Fig. 15 e 16 ). Isso levanta a questão de saber se o CTC e essas populações modernas de cães compartilham uma história comum de mistura e são descendentes das mesmas populações ancestrais.
Realizamos uma análise MixMapper que incluiu HXH na árvore do andaime e observamos que o componente europeu da CTC é extraído exclusivamente dessa população de cães alemães neolíticos primitivos ( Tabela Suplementar 13 ). Pelo contrário, os cães afegãos modernos geralmente demonstram ascendência inferida dos cães das vilas europeias modernas. Isso sugere que os cães modernos da vila afegã e o CTC são o resultado de eventos independentes de mistura.
Modelo demográfico e tempo de divergência
A composição genética distinta dos cães neolíticos europeus, em comparação com os cães europeus modernos, indica que, embora as populações antigas e contemporâneas compartilhem ancestralidade genômica substancial, é provável que algum grau de estrutura populacional esteja presente no continente. Os cães neolíticos representariam, portanto, um ramo agora extinto que é um tanto divergente do clado europeu moderno. Além disso, nosso modelo mais adequado de demografia moderna e antiga de canídeos usando ADMIXTUREGRAPH envolveu uma topologia que seria consistente com uma única linhagem de cães divergindo dos lobos ( Fig. 5a e Tabela Suplementar 14 ). Portanto, tentamos inferir o tempo de divergência de HXH e NGD de cães europeus modernos após a divergência da linhagem indiana que, de acordo com a análise de árvores de NJ, é o clado irmão do ramo da Eurásia Ocidental. Observamos que este é um modelo bifurcador simplista do que pode ter sido uma estrutura geográfica européia mais complexa e um fluxo gênico da Eurásia a longo prazo.Primeiro, realizamos uma análise G-PhoCS 25 baseada em coalescentes do modelo na Fig. 5b para obter estimativas de tempo de divergência e diversidade populacional ( Tabela Suplementar 15 e Nota Complementar 12 ). A análise foi realizada em dados de sequência de 16.434 loci de 1 kb de comprimento previamente identificados 26 . Diferentemente da análise baseada em SNP descrita acima, a chamada de genótipo de amostra única foi realizada sem um esquema específico de determinação e restringimos nossa análise a oito genomas de canídeos com cobertura variando de 8 a 24 ×. Quando incluímos apenas cães modernos, observamos que as populações de lobos pareciam divergir rapidamente, concordando com estudos anteriores 26 , 27 , enquanto a ramificação das principais linhagens de cães ocorreu durante um período de tempo muito maior. Descobrimos que o tempo de divergência cão-lobo (não calibrado) em unidades do número esperado de mutações por local (0,5247 × 10-4 ) foi semelhante ao relatado em Freedman et al . 26 ; no entanto, o tempo de divergência do nosso cão (0,2786 × 10-4 ) foi menor que o de Freedman et al . 26 , mas semelhante à Wang et al . 7 , provavelmente como resultado do uso de cães da vila do sudeste asiático em vez do dingo ( Tabela Suplementar 16 ). Também descobrimos que o tamanho efetivo da população de cães da aldeia era 5 a 10 vezes maior que o do boxeador.
Quando incluímos as amostras antigas na análise G-PhoCS, todos os tempos de divergência aumentaram acentuadamente (exceto a divisão entre cães boxer e europeus). É provável que esses resultados se devam a danos remanescentes post-mortem, inflando artificialmente a variação nas amostras antigas e alongando os comprimentos dos galhos na análise G-PhoCS, pois detectamos um excesso de variantes privadas nas três amostras antigas em comparação com os cães da aldeia europeus. Por isso, criamos um novo método para estimar o tempo de divisão HXH / NGD-Europeu (τ 1 ) utilizando os resultados G-PhoCS apenas para amostras modernas e que seria robusto a vieses resultantes do uso de amostras antigas ( Nota Suplementar 13 e Nota Suplementar) Fig. 24) Especificamente, calculamos a quantidade relativa observada de compartilhamento de alelo derivado exclusivo para cães da aldeia europeus e HXH / NGD versus aquele exclusivo para cães da vila europeus e indianos. As duas principais vantagens dessa estimativa são que (a) depende apenas de locais variáveis descobertos anteriormente em cães modernos com maior cobertura (nosso chamado de genótipo em amostras antigas provavelmente será muito mais preciso nessas situações) e (b) usa apenas um único cromossomo de cada população (que pode ser escolhido aleatoriamente) e, portanto, não requer a chamada exata de heterozigotos (isto é, não deve ser sensível à menor cobertura de nossas amostras antigas). Como esperado, os cães europeus compartilham mais alelos derivados com os cães antigos do que os cães da aldeia indiana, com proporções de 1.186-1.217 para HXH e 1.195-1.231 para NGD (Tabela Suplementar 17 ).
Em seguida, calculamos a expectativa dessa relação usando a teoria coalescente e iteramos sobre os possíveis valores de τ 1 até que a expectativa da relação caísse no intervalo de confiança observado. Embora nossas estimativas de tempos de divergência sejam em unidades do número esperado de mutações, podemos usar a idade de nossas amostras antigas para calibrar o tempo de divergência resultante em anos. Utilizamos a idade da amostra de HXH para definir um limite superior para a taxa de mutação anual μ , pois a amostra deve ser menor do que o tempo em anos desde a divergência de HXH e cães europeus modernos. Dado que a amostra tem ∼ 7.000 anos, inferimos que um limite superior para μ é 5,6 × 10 −9por geração (assumindo um tempo de geração de 3 anos, com um IC de 95% para o limite superior de 3,7 × 10 a 9,4 a 7,4 × 10 a 9 , Figura suplementar 25 ). Esse limite superior, que representa a maior taxa de mutação potencialmente compatível com a idade de nossas amostras, é consistente com a taxa de μ = 4 × 10 −9 por geração sugerida por Skoglund et al . 28 e Frantz et al . 12 , duas taxas também calibradas por amostras antigas. Quando calibramos τ 1 usando essa taxa de mutação, estimamos um valor de of 6.500 a 12.900 anos para HXH e ∼ 6.400 a 12.600 anos para a DNG.
A partir da análise do G-PhoCS, estimamos ainda que os cães modernos europeus e indianos divergiram de 13.700 a 17.900 anos atrás, os quais divergiram dos cães do Sudeste Asiático entre 17.500 a 23.900 anos como um evento de divergência de cães basal. Finalmente, estimamos o tempo de divergência cão-lobo em 36.900 a 41.500 anos atrás ( Fig. 5b ). Observamos que, embora em consonância com estudos anteriores 7 , 26 , nossas estimativas de divergência entre cães leste-oeste são muito mais antigas do que as relatadas em Frantz et al . 12 (6.000 a 14.000 anos atrás). Enquanto usamos uma abordagem bayesiana com G-PhoCS para inferir tempos de divergência, Frantz et al . 12confiam na abordagem múltipla sequencialmente coalescente de Markov (MSMC), cujo desempenho depende fortemente da precisão das fases genômicas 29 .
Variantes funcionais associadas à domesticação
Como resultado do processo de domesticação, porções específicas dos genomas de cães se diferenciaram significativamente dos lobos 30 . Para determinar o status de domesticação dos três cães neolíticos, avaliamos a diversidade de haplótipos nos locais de domesticação candidatos. Usando apenas cães e lobos da raça, um estudo anterior identificou 36 locais candidatos à domesticação 30 ( Tabela Suplementar 18 ). No entanto, nossa análise de um conjunto de amostras mais diversificado, que inclui cães da aldeia, confirma apenas 18 desses locais como alvos putativos de domesticação, o restante provavelmente está associado à formação de raças ( Tabela Suplementar 19 e Nota Suplementar 14) O HXH parecia homozigoto para o haplótipo tipo cachorro, exceto um desses 18 loci, e, portanto, era muitas vezes indistinguível da maioria dos cães modernos. O NGD mais jovem parecia um cão, exceto dois loci. A CTC, no entanto, era heterozigótica para o haplótipo semelhante ao lobo em seis locais, compatível com o aumento da ancestralidade do lobo descrito acima.O Neolítico viu mudanças drásticas na cultura e no comportamento humano, incluindo o advento da agricultura, resultando em uma mudança para dietas mais ricas em amido. O número elevado de cópias de AMY2B , que está associado ao aumento da eficiência do metabolismo do amido, tem sido frequentemente sugerido como uma forte característica candidata à domesticação, embora se saiba que o número de cópias de AMY2B varia amplamente em diversas coleções de lobos modernos e cães de raça 26 , 31 , 32 . Embora o haplótipo canino esteja presente nas três amostras neolíticas nesse locus ( Fig. 6a ), nenhuma mostrou evidência da expansão extrema do número de cópias do AMY2B ( Fig. 6b) Com base na profundidade da leitura, estimamos que o CTC e o HXH tenham duas cópias do gene AMY2B, enquanto o NGD possuiu três cópias, e não duas, conforme relatado anteriormente 12 ( Nota Complementar 14 ). A análise do conjunto completo de amostras de caninos mostra uma distribuição bimodal do número de cópias, com a maioria dos cães modernos com> 6 cópias AMY2B , enquanto poucos carregam 2 ou 3 cópias 32 . Presume-se que esse aumento dinâmico e extremo do número de cópias seja o resultado de uma expansão tandem do gene AMY2B 30 . Outras análises de perfis NGD leitura profundidade revelou a presença de uma maior, ~ 2 megabases englobando a duplicação segmentar amy2blocus gênico no cromossomo 6 e se estendendo proximalmente em direção ao centrômero ( Fig. 26 ). Essa duplicação está presente em 11 das amostras modernas de cães analisadas e parece ser independente da expansão extrema do número de cópias do próprio gene AMY2B ( Nota Complementar 14 ).
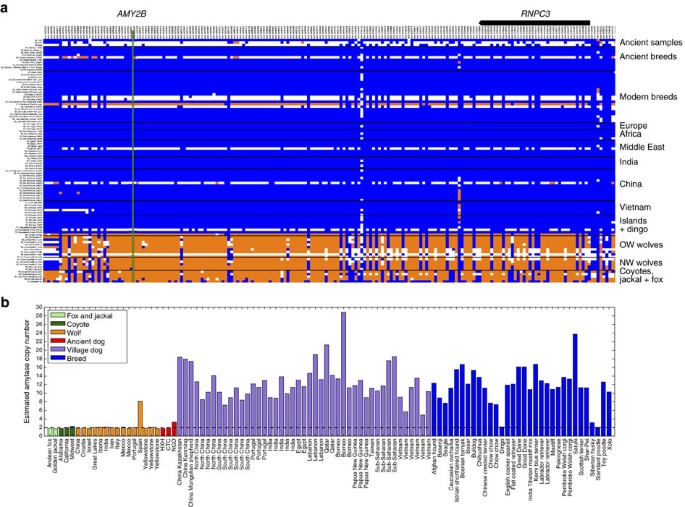
( Um ) Genótipo matriz de locais seleccionados dentro F ST derivada de domesticação locus de 12 (CHR6: 46.854.109-47.454.177) 30
. Os genótipos SNP são representados como homozigotos para o alelo de
referência (0/0; azul), heterozigotos (0/1; branco) ou homozigotos (1/1;
laranja) para o alelo alternativo. As posições de AMY2B (linha verde) e RNPC3 (modelo acima) são indicadas. ( b ) Estimativa com base na leitura do número de cópias AMY2B
para a raposa andina (verde claro), chacal dourado (verde claro),
coiotes (verde escuro), lobos (laranja), amostras antigas (vermelho),
cães da vila (roxos) ) e cães de raças (azuis). A linha tracejada indica
o número de cópias diplóides de dois.
Discussão
Este
estudo paleogenômico fornece várias novas idéias sobre a história da
domesticação de cães na Europa. Encontramos fortes evidências de
continuidade genética do paleolítico para o neolítico e, até certo
ponto, para o presente. Além disso, não encontramos evidências de que
uma população de cães paleolíticos europeus agora extinta contribua para
uma população de cães geneticamente distinta do neolítico inicial ou
final e, portanto, nossos resultados não suportam a hipótese de uma
grande substituição populacional do leste da Ásia durante nesta era. Em
vez disso, descobrimos que o NGD é geneticamente muito semelhante ao
HXH, com ambos possuindo ∼70
a 80% de ascendência européia moderna. Além disso, o CTC provavelmente
descende diretamente de uma população representada pelo HXH, apontando
alguma continuidade genética em todo o Neolítico (mais de 2.000 anos) na
Europa Central.
No entanto, os eventos de mistura observados em cães neolíticos europeus, mas não na maioria dos cães modernos (e mesmo assim em menor grau) da mesma região sugerem algum grau de estrutura populacional no continente durante esse período. Isso é refletido ainda mais por HXH e NGD portando ancestralidade do Sudeste Asiático, mas sem a ancestralidade compartilhada entre a CTC e os modernos cães de vila do Oriente Médio, Central e Sul da Ásia, mesmo que a NGD e a CTC sejam contemporâneas (4.800 e 4.700 anos, respectivamente). É provável que, nesse cenário de estrutura populacional, uma subpopulação distinta da de HXH, CTC e NGD tenha se tornado dominante nos cães europeus modernos, o que pode explicar a mudança observada no mtDNA do haplogrupo C para A, especialmente se essa subpopulação também passou por um forte gargalo.Um apoio adicional à estrutura da população vem do agrupamento de todas as amostras antigas dentro de C1 em um sub-haplogrupo distinto do dos cães modernos, enquanto também é digno de nota que os haplogrupos não-C, incluindo A, são mais aparentes no sudeste da Europa no registro arqueológico12 .
A CTC mostra padrões de mistura semelhantes às populações modernas de cães da Ásia Central e do Oriente Médio. Considerando que a idade das amostras fornece um período de tempo (entre 7.000 e 5.000 anos atrás) para a CTC obter seu componente ancestral exclusivo, e que o crânio foi encontrado ao lado de dois indivíduos associados à cultura Neolithic Corded Ware, especulamos que isso Esse componente foi derivado das populações de cães que acompanhavam as estepes migrando do leste 13 .
Análises que incorporam mistura em seu modelo mostram uma proporção significativa de ascendência indiana moderna na CTC. No entanto, além disso, existe um potencial componente semelhante ao lobo observado em nossas análises NGSadmix e Spacemix, além de um componente do sudeste asiático que aparece nos três cães neolíticos. Dado um quadro tão complexo da mistura, com quatro fontes potenciais que devem ser inferidas a partir de um único genoma, talvez não seja surpreendente que métodos diferentes demonstrem variabilidade em suas proporções de mistura inferidas do tipo indiano (de 25% no NGSadmix até 69% no ADMIXTUREGRAPH ) Esperamos que mais genomas da Europa Central desta época ajudem a esclarecer esse quadro complicado de mistura no futuro.
Nossa estimativa mais antiga do tempo de divergência "leste-oeste" de 17.500 a 23.900 anos atrás nega a necessidade de invocar uma hipótese de origem dupla de cães sugerida por Frantz et al . 12 . A continuidade genômica vemos entre a nossa 7.000 anos hxh, a ~ 5.000 anos NGD e amostras europeias modernas implica que, se houvesse qualquer tipo de reposição da população, que deve ter ocorrido antes do Neolítico (e talvez muito mais cedo dado o matrilinear continuidade entre HXH, CTC e Bona – Oberkassel). Porém, diferentemente das proposições de Frantz et al . 12, essa substituição seria necessariamente independente da rotatividade observada no mtDNA das linhagens C-A, pois parece ter ocorrido pelo menos 2.000 anos após o final do neolítico (ou seja, separado por pelo menos 4.000 anos).
Também estimamos o tempo de divergência cão-lobo em 36.900 a 41.500 anos ( Fig. 5b ), o que é consistente com as previsões do antigo genoma do lobo Taimyr 28 . Como a domesticação deve ter ocorrido subseqüentemente à divergência cão-lobo e antes da divergência cão do Sudeste Asiático ( ∼ 17.500–23.900 anos atrás; Fig. 5b ), nossos resultados fornecem um limite superior e inferior para o início da domesticação do cão, entre ∼ 20.000 e 40.000 anos atrás. Até o momento, o Sudeste Asiático, a Europa, o Oriente Médio e a Ásia Central foram propostos como possíveis locais para a origem da domesticação de cães com base em dados genômicos modernos, evidências arqueológicas e linhagens mitocondriais antigas 5 ,7 , 9 , 33 . Embora nossas análises de três genomas neolíticos da Europa tenham ajudado a restringir o tempo da domesticação, eles não têm idade suficiente nem a ampla distribuição geográfica necessária para resolver esse debate. No entanto, nosso trabalho deixa claro que a estrutura e a mistura populacional têm sido uma característica proeminente da evolução canina por um período substancial de tempo. É pouco provável que as análises genéticas populacionais baseadas apenas ou principalmente em dados modernos sejam responsáveis por essa complexidade ao modelar a história demográfica de cães e, portanto, os dados paleogenômicos dos restos do Paleolítico Superior em toda a Eurásia serão cruciais para finalmente resolver os locais da domesticação de cães.
A digestão aprimorada do amido através da expansão extrema do número de cópias AMY2B foi postulada como uma adaptação à mudança da dieta carnívora dos lobos para a dieta rica em amido dos cães domésticos 30 . Embora nenhuma das amostras neolíticas alemãs possua a expansão do número de cópias do gene AMY2B associado à digestão do amido, descobrimos que esse gene está presente em três cópias no NGD, embora isso se deva a uma grande duplicação segmentar que é compartilhada com vários cães modernos , um evento separado das duplicações em tandem do AMY2B . Isso sugere que a seleção inicial nesse locus pode ter sido conduzida de forma independente por algum outro fator que não o número de cópias AMY2B . A ausência do extremoO aumento do número de cópias do AMY2B nessas amostras antigas indica que a varredura seletiva associada à expansão do AMY2B deve ter ocorrido bem após o advento da agricultura e do neolítico na Europa. Isso é consistente com as descobertas recentes de que o número de cópias do AMY2B é mais alto nas populações modernas de cães originários de regiões geográficas com sociedades agrárias pré-históricas e mais baixo nas regiões onde os seres humanos não confiavam na agricultura para subsistência 34 e apóia a alegação de que a expansão ocorreu após a domesticação inicial (possivelmente após a migração de dingoes para a Austrália há 3.500 a 5.000 anos) 34. Um padrão semelhante foi observado em humanos, onde os alelos associados à persistência de lactase na Europa não atingiram freqüências significativas até pelo menos a Idade do Bronze, ou seja, 3.000 anos após a introdução da pecuária pastoral 35 .
No geral, nossas descobertas revelam uma história de cães domésticos tão intricada quanto a das pessoas com quem eles viviam. A inferência de padrões complexos de fluxo gênico é desafiadora ou até impossível quando apenas amostras modernas são estudadas. Portanto, a aquisição de um conjunto mais amplo de amostras antigas, incluindo antigos representantes da Ásia Central e do Sudeste e do Oriente Médio, será crucial para esclarecer melhor os detalhes da domesticação e evolução dos cães.
No entanto, os eventos de mistura observados em cães neolíticos europeus, mas não na maioria dos cães modernos (e mesmo assim em menor grau) da mesma região sugerem algum grau de estrutura populacional no continente durante esse período. Isso é refletido ainda mais por HXH e NGD portando ancestralidade do Sudeste Asiático, mas sem a ancestralidade compartilhada entre a CTC e os modernos cães de vila do Oriente Médio, Central e Sul da Ásia, mesmo que a NGD e a CTC sejam contemporâneas (4.800 e 4.700 anos, respectivamente). É provável que, nesse cenário de estrutura populacional, uma subpopulação distinta da de HXH, CTC e NGD tenha se tornado dominante nos cães europeus modernos, o que pode explicar a mudança observada no mtDNA do haplogrupo C para A, especialmente se essa subpopulação também passou por um forte gargalo.Um apoio adicional à estrutura da população vem do agrupamento de todas as amostras antigas dentro de C1 em um sub-haplogrupo distinto do dos cães modernos, enquanto também é digno de nota que os haplogrupos não-C, incluindo A, são mais aparentes no sudeste da Europa no registro arqueológico12 .
A CTC mostra padrões de mistura semelhantes às populações modernas de cães da Ásia Central e do Oriente Médio. Considerando que a idade das amostras fornece um período de tempo (entre 7.000 e 5.000 anos atrás) para a CTC obter seu componente ancestral exclusivo, e que o crânio foi encontrado ao lado de dois indivíduos associados à cultura Neolithic Corded Ware, especulamos que isso Esse componente foi derivado das populações de cães que acompanhavam as estepes migrando do leste 13 .
Análises que incorporam mistura em seu modelo mostram uma proporção significativa de ascendência indiana moderna na CTC. No entanto, além disso, existe um potencial componente semelhante ao lobo observado em nossas análises NGSadmix e Spacemix, além de um componente do sudeste asiático que aparece nos três cães neolíticos. Dado um quadro tão complexo da mistura, com quatro fontes potenciais que devem ser inferidas a partir de um único genoma, talvez não seja surpreendente que métodos diferentes demonstrem variabilidade em suas proporções de mistura inferidas do tipo indiano (de 25% no NGSadmix até 69% no ADMIXTUREGRAPH ) Esperamos que mais genomas da Europa Central desta época ajudem a esclarecer esse quadro complicado de mistura no futuro.
Nossa estimativa mais antiga do tempo de divergência "leste-oeste" de 17.500 a 23.900 anos atrás nega a necessidade de invocar uma hipótese de origem dupla de cães sugerida por Frantz et al . 12 . A continuidade genômica vemos entre a nossa 7.000 anos hxh, a ~ 5.000 anos NGD e amostras europeias modernas implica que, se houvesse qualquer tipo de reposição da população, que deve ter ocorrido antes do Neolítico (e talvez muito mais cedo dado o matrilinear continuidade entre HXH, CTC e Bona – Oberkassel). Porém, diferentemente das proposições de Frantz et al . 12, essa substituição seria necessariamente independente da rotatividade observada no mtDNA das linhagens C-A, pois parece ter ocorrido pelo menos 2.000 anos após o final do neolítico (ou seja, separado por pelo menos 4.000 anos).
Também estimamos o tempo de divergência cão-lobo em 36.900 a 41.500 anos ( Fig. 5b ), o que é consistente com as previsões do antigo genoma do lobo Taimyr 28 . Como a domesticação deve ter ocorrido subseqüentemente à divergência cão-lobo e antes da divergência cão do Sudeste Asiático ( ∼ 17.500–23.900 anos atrás; Fig. 5b ), nossos resultados fornecem um limite superior e inferior para o início da domesticação do cão, entre ∼ 20.000 e 40.000 anos atrás. Até o momento, o Sudeste Asiático, a Europa, o Oriente Médio e a Ásia Central foram propostos como possíveis locais para a origem da domesticação de cães com base em dados genômicos modernos, evidências arqueológicas e linhagens mitocondriais antigas 5 ,7 , 9 , 33 . Embora nossas análises de três genomas neolíticos da Europa tenham ajudado a restringir o tempo da domesticação, eles não têm idade suficiente nem a ampla distribuição geográfica necessária para resolver esse debate. No entanto, nosso trabalho deixa claro que a estrutura e a mistura populacional têm sido uma característica proeminente da evolução canina por um período substancial de tempo. É pouco provável que as análises genéticas populacionais baseadas apenas ou principalmente em dados modernos sejam responsáveis por essa complexidade ao modelar a história demográfica de cães e, portanto, os dados paleogenômicos dos restos do Paleolítico Superior em toda a Eurásia serão cruciais para finalmente resolver os locais da domesticação de cães.
A digestão aprimorada do amido através da expansão extrema do número de cópias AMY2B foi postulada como uma adaptação à mudança da dieta carnívora dos lobos para a dieta rica em amido dos cães domésticos 30 . Embora nenhuma das amostras neolíticas alemãs possua a expansão do número de cópias do gene AMY2B associado à digestão do amido, descobrimos que esse gene está presente em três cópias no NGD, embora isso se deva a uma grande duplicação segmentar que é compartilhada com vários cães modernos , um evento separado das duplicações em tandem do AMY2B . Isso sugere que a seleção inicial nesse locus pode ter sido conduzida de forma independente por algum outro fator que não o número de cópias AMY2B . A ausência do extremoO aumento do número de cópias do AMY2B nessas amostras antigas indica que a varredura seletiva associada à expansão do AMY2B deve ter ocorrido bem após o advento da agricultura e do neolítico na Europa. Isso é consistente com as descobertas recentes de que o número de cópias do AMY2B é mais alto nas populações modernas de cães originários de regiões geográficas com sociedades agrárias pré-históricas e mais baixo nas regiões onde os seres humanos não confiavam na agricultura para subsistência 34 e apóia a alegação de que a expansão ocorreu após a domesticação inicial (possivelmente após a migração de dingoes para a Austrália há 3.500 a 5.000 anos) 34. Um padrão semelhante foi observado em humanos, onde os alelos associados à persistência de lactase na Europa não atingiram freqüências significativas até pelo menos a Idade do Bronze, ou seja, 3.000 anos após a introdução da pecuária pastoral 35 .
No geral, nossas descobertas revelam uma história de cães domésticos tão intricada quanto a das pessoas com quem eles viviam. A inferência de padrões complexos de fluxo gênico é desafiadora ou até impossível quando apenas amostras modernas são estudadas. Portanto, a aquisição de um conjunto mais amplo de amostras antigas, incluindo antigos representantes da Ásia Central e do Sudeste e do Oriente Médio, será crucial para esclarecer melhor os detalhes da domesticação e evolução dos cães.
Métodos
Antecedentes arqueológicos
Para a amostra HXH, um único osso petroso foi identificado na estrutura interna da vala de Herxheim, um local neolítico precoce na Alemanha descoberto em 1996, que continha material arqueológico da cultura Linearbandkeramik. Herxheim contém uma quantidade significativa de restos de fauna, incluindo> 250 restos de cães que constituem a maior série óssea de cães neolíticos primitivos da Europa Ocidental. A 14 C datado de 5.223-5.040 cal. O BCE (95,4%) foi estimado para o osso (Mams-25941: 6186 ± 30, calibrado com OxCal 4.2 (ref. 36 ) usando a curva de calibração IntCal13 37 ).Para a amostra da CTC, todo o crânio de um cão foi encontrado na Kirschbaumhöhle (Cherry Tree Cave) em Franconian Alb, Alemanha 14 ( Fig. 27 ). A caverna foi descoberta em 2010 e contém restos humanos e animais de pelo menos seis períodos pré-históricos. O CTC era um cão adulto que demonstrava semelhança morfológica com o chamado Torfhund ( Canis familiaris palustris ) e foi encontrado perto de dois crânios humanos datados do início do Neolítico Final (2.800–2.600 cal. AEC). A 14 C datado de 2.900-2.632 cal. O BCE (95,4%) foi estimado para o crânio (Erl-18378: 4194 ± 45, calibrado com OxCal 4.2 usando a curva de calibração IntCal13). Consulte a Nota Suplementar 1 para obter mais detalhes.
Isolamento e triagem de DNA
Para a amostra HXH, a parte petrosa do osso temporal da amostra HXH foi preparada em instalações de salas limpas dedicadas ao DNA antigo no Trinity College Dublin (Irlanda). A extração do DNA foi realizada usando o método da coluna Silica, como descrito em MacHugh et al . 38 . Duas bibliotecas genômicas foram preparadas como descrito em Gamba et al . 39 . A triagem de uma biblioteca por meio de uma execução do Illumina MiSeq e o mapeamento em relação a vários genomas de referência demonstraram que as leituras para esta amostra foram mapeadas quase exclusivamente para o genoma do CanFam3, revelando que era um canídeo. Controles em branco foram utilizados por toda parte. Ver Nota Suplementar 2 , Figs. 28 e 29 Suplementares eTabelas suplementares 20 e 21 para mais detalhes.Para a CTC, uma ampla preparação foi realizada em instalações dedicadas de DNA antigas do Grupo Paleogenético da Johannes Gutenberg-University Mainz sob regras estritas para a prevenção de contaminação, como descrito em Bramanti et al . 40 . O DNA foi extraído independentemente duas vezes do osso petroso usando um protocolo fenol-clorofórmio 41 . Um total de quatro bibliotecas genômicas com índice duplo foi preparado como descrito em Hofmanová et al . 17 . Uma biblioteca foi rastreada quanto ao conteúdo de DNA endógeno via sequenciamento Illumina MiSeq, com 61,5% das leituras mapeadas para o CanFam3. Controles em branco foram utilizados por toda parte. Veja a Nota Suplementar 3 , Fig. 30e Tabela Suplementar 22 para mais detalhes.
Sequenciamento de genoma e processamento bioinformático
Combinações de várias bibliotecas genômicas de cada amostra antiga (CTC e HXH) foram sequenciadas em duas faixas de um Illumina HiSeq 2500 1TB no New York Genome Center (NYGC) usando o modo de execução de alto rendimento para produzir leituras de 2 × 125 pb em pares . As leituras foram cortadas, mescladas e filtradas usando uma versão modificada do antigo protocolo de DNA descrito por Kircher 42 . As leituras mescladas foram então mapeadas usando o BWA aln 43 para uma versão modificada do genoma de referência CanFam3.1 contendo um cromossomo Y. As leituras duplicadas foram identificadas e marcadas usando PICARD MarkDuplicates, resultando em uma cobertura média para ambas as amostras de 9 ×. Além disso, a cobertura média para os cromossomas X e Y foi ~5 × para ambas as amostras, indicando que eram do sexo masculino. O comprimento médio do fragmento para ambas as amostras variou de 60 a 70 pb. Os efeitos da degradação post-mortem foram avaliados usando o MapDamage_v1.0 (ref. 15 ), revelando extensos danos em 5 ′ C> T e 3 ′ G> A. As leituras de terminação única para NGD extraídas de um arquivo BAM contendo todas as leituras mapeadas foram processadas usando o mesmo pipeline. Veja a Nota Suplementar 4 para mais detalhes.A estimativa da probabilidade do genótipo e a chamada do genótipo para todas as três amostras antigas foram realizadas usando um chamador personalizado que leva em consideração os padrões de danos pós-morte identificados pelo MapDamage com base no modelo descrito em Hofmanová et al . 17 . Resumidamente, os padrões de dano em relação à posição de leitura são adequados a uma distribuição Weibull da forma a × exp (- ( x c ) × b ), em que x é a proporção de bases C> T ou G> A danificadas em uma posição específica ao longo da leitura (diferentemente de Hofmanová et al . 17 , encontramos um ajuste um pouco melhor com um Weibull do que quando assumimos decaimento exponencial) ( Fig. 31 complementar )) Qualquer site com cobertura <7 30="" a="" al="" ausente.="" c="" como="" de="" deve="" digo="" dispon="" disso="" em="" escala="" escolhido.="" est="" foi="" font="" gen="" heterozigoto="" homozigoto="" m="" maior="" nima="" o="" ou="" phred="" posi="" pr="" probabilidade="" qualidade="" qualquer="" que="" relatado="" seja="" ter="" tipo="" um="" uma="" vel="" ximo="">https://github.com/kveeramah/aDNA_GenoCaller . Esse protocolo diminuiu substancialmente a super-representação dos locais C> T e G> A identificados pelo GATK UnifiedGenotyper 44 , que não são responsáveis por danos post-mortem. Além disso, as chamadas de base com índice de qualidade <15 a="" as="" base="" chamada="" chamadas="" com="" das="" de="" durante="" e="" foram="" gen="" inclu="" leituras="" mapeamento="" n="" ndice="" o="" qualidade="" tipo.="" um=""> 40 (que pode ocorrer durante a mesclagem de leitura na extremidade emparelhada) foram ajustadas para 40. Consulte a Nota Suplementar 5 eFigs. 32 e 33 complementares para mais detalhes.
Conjunto de dados de referência
Para construir um conjunto de dados da sequência do genoma, além das três amostras antigas, examinamos os dados da sequência do genoma inteiro de 96 canídeos modernos. Genomas adicionais foram gerados usando o seqüenciamento de Illumina para um dinamarquês e lobo ibérico (SRP073312). Também postamos leituras de sequenciamento no SRA para um cão de vila português, cão de pastor da Mongólia chinesa e um cachorro de vila da África Subsaariana (SRP034749). Todos os dados restantes do genoma foram adquiridos a partir de conjuntos de dados publicados anteriormente depositados no SRA. Como acima, as leituras de todos os canídeos modernos foram alinhadas ao CanFam3.1 usando BWA, seguidas pela recalibração do escore de qualidade do GATK e chamada de genótipo usando o HaplotypeCaller 44 . Esses dados foram complementados com dados de genótipo para seis canídeos de Freedman et al . 26(basenji, dingo, chacal dourado, lobo croata, lobo israelense e lobo chinês). Geramos três conjuntos de chamadas diferentes com diferentes esquemas de apuração. O conjunto de chamadas 1 inclui todas as variantes dos genomas antigo e contemporâneo, representando o conjunto mais abrangente de variantes, mas pode mostrar vieses devido a diferenças na cobertura entre os conjuntos de amostras. O conjunto de chamadas 2 inclui apenas variantes descobertas nos três genomas antigos. O conjunto de chamadas 3 inclui apenas sites descobertos como variáveis nos lobos do Novo Mundo e é o principal conjunto de chamadas utilizado para a maioria das análises. Consulte a Nota Suplementar 6 para obter mais detalhes.Para construir um conjunto de dados de matriz SNP, foram obtidos conjuntos de dados de matriz SNP canina de Shannon et al . 9 e Pilot et al . 18 . Os genótipos também foram suplementados por dados dos seis canídeos relatados em Freedman et al . 26 .
Análise estatística
A profundidade média de sequenciamento para o mtDNA foi de 179 ×, 208 × e 170 × nas amostras CTC, HXH e NGD, respectivamente. Amostras antigas de seqüências de consenso de mtDNA foram alinhadas ao alinhamento canídeo de Thalmann et al . 5 , que contêm genomas completos de mtDNA para os canídeos modernos e antigos. Uma árvore NJ foi construída com um modelo de substituição TN93 (500 bootstraps) usando MEGA 6.06 (ref. 45 ). Uma outra árvore NJ foi construída com amostras adicionais de C1 e C2 de Duleba et al . 19Veja a Nota Suplementar 7 para mais detalhes.As árvores NJ foram construídas para o conjunto SNP de todo o genoma usando o pacote macaco R 46 usando matrizes de distância baseadas na métrica da divergência de sequência de Gronau et al . 25 . Cem réplicas de bootstrap foram geradas dividindo o genoma em janelas de 5 cM e amostrando com substituição para determinar o suporte do nó. Veja a Nota Suplementar 8 para mais detalhes.
A PCA foi realizada no conjunto de dados da matriz SNP e no conjunto de chamadas SNP do genoma 3 usando smartpca, parte do pacote EIGENSOFT versão 3.0 (ref. 47 ). Tanto o genótipo diplóide quanto o pseudo-haploide chamam com e sem SNPs C <> T e G <> A (os locais mais prováveis de sofrer danos post-mortem) foram usados para construir a PCA, mas pouca diferença foi encontrada entre essas análises. O SpaceMix 24 foi usado para criar um mapa geogenético e inferir possíveis eventos de mistura de longa distância através deste mapa usando os dados da matriz SNP, permitindo apenas SNPs separados por pelo menos 100 kb e não mais de cinco indivíduos por população. Foram executadas várias execuções com 10 burn-ins iniciais de 100.000 gerações e um final longo de 10.000.000 gerações. ADMIXTURA (v. 1.22) 48foi usado para realizar uma análise de cluster não supervisionada nos dados da matriz SNP dos cães antigos e um subconjunto de 105 cães modernos que forneciam uma representação global da estrutura do cão, enquanto o NGSadmix 49 foi usado para realizar uma análise semelhante para os dados do genoma do SNP enquanto tomava consideração a incerteza do genótipo, examinando as probabilidades do genótipo. Validação cruzada foi realizada para a análise ADITIVO para identificar o número mais adequado de clusters, K . Consulte a Nota Suplementar 9 e as Figuras Suplementares 34 e 45 para obter mais detalhes.
As estatísticas do subgrupo- f3 foram usadas para avaliar a deriva genética relativa entre cães antigos e modernos. Esse método foi usado anteriormente em estudos antigos de DNA para investigar como as populações modernas estão geneticamente relacionadas a uma amostra antiga 17 , 21 , 35 , 50. Supondo um modelo simples de três populações sem fluxo gênico pós-divergência, em que a população C é um subgrupo para A e B, o valor dessa estatística refletirá a quantidade de desvio compartilhado entre A e B em relação a C. Se uma população (por exemplo, B) é mantido constante; nesse caso, um cão antigo, e a introdução de populações diferentes para representar A fornecerá estimativas relativas de similaridade genética com B (observe que isso não faz suposições com relação à complexidade da história demográfica que conecta as populações A e B). Mapas estatísticos do grupo externo f3 foram criados usando os pacotes R ggplot2 e mapas usando o conjunto de dados Natural Earth de domínio público ( http://www.naturalearthdata.com ). Dforam utilizadas estatísticas para identificar potenciais testes de mistura antiga cão-lobo e razão f4 para estimar as proporções de mistura cão-cão e cão-lobo foram calculadas usando o Admixtools 22 . Consulte a Nota Suplementar 10 e a Fig. 46 Suplementar para obter mais detalhes. Tanto o MixMapper 23 quanto o ADMIXTUREGRAPH 22foram utilizados para realizar inferência baseada em modelos de eventos específicos de mistura envolvendo os três cães antigos. O MixMapper foi realizado no conjunto de dados SNP e no conjunto do genoma SNP, enquanto ADMIXTUREGRAPH foi realizado apenas no conjunto do conjunto de dados do genoma. A significância foi avaliada usando um procedimento de canivete de bloco ponderado para todos os cinco tipos de análise. As posições dos mapas genéticos para cada SNP usado nessas análises foram inferidas por Auton et al . 51Consulte a Nota Suplementar 11 e as Figuras Suplementares 47 e 48 para obter mais detalhes.
O G-PhoCS 25 foi usado para estimar tempos de divergência, tamanhos efetivos de população e taxas de migração para várias combinações modernas de cães e lobos, usando alinhamentos de sequências de 16.434 locos 'neutros' previamente identificados em Freedman et al . 26após o LiftOver de CanFam3 para CanFam3.1. Árvores NJ foram construídas para informar a topologia da divergência populacional. Um total de 500.000 iterações de Monte Carlo da cadeia Markov (MCMC) foi considerado suficiente para convergência para nossos dados, com os últimos 200.000 usados para estimar distribuições posteriores. Em seguida, desenvolvemos uma abordagem numérica baseada na teoria coalescente para prever a proporção de locais derivados compartilhados entre HXH / NGD e cães de vila europeus versus cães de vila indianos e europeus, devido a um tempo de divergência específico de HXH / NGD em unidades de número esperado de mutações ( P 1 ) Nossa expectativa foi condicionada às seguintes estimativas de parâmetros dos G-PhoCs: N e para a população ancestral européia / boxer ( θ 1 ),N e para população ancestral europeia / indiana ( θ 2 ), tempo de divergência para a Europa e Boxer ( P 0 ), tempo de divergência para Europa / Índia ( P 2 ) e tempo de divergência para Europa-Índia / Ásia ( P 3 ) bem como a porcentagem de HXH que é composta pela mistura asiática ( α ) da análise da razão f4 ( Tabela Suplementar 23) Intervalos de confiança foram estimados por reamostragem dos parâmetros G-PhoCS de suas distribuições posteriores e encontrando razões de compartilhamento de alelos derivadas previstas que estavam dentro de um intervalo determinado para os dados observados por uma abordagem de reamostragem ponderada de canivetes. Consulte as Notas Suplementares 12 e 13 , as Figuras Suplementares 49 a 55 e a Tabela Suplementar 23 para obter mais detalhes.
Coordenadas de 30 'locais de domesticação' putativos foram obtidas de Axelsson et al . 30 e passou das coordenadas CanFam2.0 para CanFam3.1. O SNPs do conjunto de chamadas 1 em cada janela foi extraído das amostras antigas e do nosso conjunto de dados da sequência do genoma. Formatos de arquivo de genótipo Eigenstrat foram gerados por janela usando convertf do pacote EIGENSOFT 52 e scripts personalizados foram usados para converter os arquivos de genótipo em formatos de matriz para visualização usando matrix2png 53usando um subconjunto filtrado de SNPs (frequências alélicas menores entre 0,05 e 0,49) para facilitar a visualização das matrizes. As árvores NJ foram estimadas para cada janela com o conjunto completo de SNP usando os mesmos métodos que a estimativa de árvores do genoma inteiro (veja acima). No total, os haplótipos das três amostras antigas foram classificados como cão ou lobo, por 18 matrizes que mostraram clara distinção entre haplótipos de cães e canídeos selvagens, com base na contagem média de alelos de referência calculada por janela. Consulte a Nota Suplementar 14 e as Figuras Suplementares 56–50 para obter mais detalhes.
O número de cópias genômicas no locus da amilase 2B foi estimado a partir da profundidade de leitura, conforme descrito anteriormente 54 , 55 . Especificamente, as leituras foram divididas em fragmentos de 36 pb não sobrepostos e mapeadas para uma versão com máscara repetida da referência CanFam3.1 usando o mrsFAST 56 , retornando todos os canais de leitura com duas ou menos substituições. As profundidades de leitura brutas foram tabuladas em cada posição e uma correção de perda para o conteúdo local do GC foi calculada utilizando regiões de controle não identificadas anteriormente como variável de número de cópias. A profundidade média em janelas de 3 kb foi então calculada e convertida no número estimado de cópias com base na profundidade nas regiões de controle autossômico. Consulte a Nota Suplementar 14 para obter mais detalhes.
A PCA, o tempo de divergência entre cães orientais e ocidentais e os modelos demográficos testados com ADMIXTUREGRAPH são comparados com Frantz et al . 12 e discutido na nota complementar 15 .
Disponibilidade de dados
Os dados de sequenciamento estão disponíveis no banco de dados do arquivo de leitura de sequência NCBI (SRA) sob os números de acesso SRS1407451 (CTC) e SRS1407453 (HXH), e os genomas mitocondriais estão disponíveis no GenBank sob os acessos KX379528 e KX379529 , respectivamente. O código gerado para chamar variantes nas amostras antigas está disponível em: https://github.com/kveeramah/aDNA_GenoCaller .Informação adicional
Como citar este artigo: Botigué, LR et al . Os genomas de cães europeus antigos revelam continuidade desde o início do neolítico. Nat. Comum. 8, 16082 doi: 10.1038 / ncomms16082 (2017).
Nota do editor: A Springer Nature permanece neutra em relação a reivindicações jurisdicionais em mapas publicados e afiliações institucionais.
Nota do editor: A Springer Nature permanece neutra em relação a reivindicações jurisdicionais em mapas publicados e afiliações institucionais.
Códigos de adesão
Referências
- 1 Parker, HG et al. Estrutura genética do cão doméstico de raça pura. Science 304 , 1160-1164 (2004).
- 2 Benecke, N. Estudos sobre restos de cães no norte da Europa. J. Archaeol. Sci. 14 , 31-49 (1987).
- 3 Perri, A. Um lobo em roupas de cachorro: domesticação inicial do cão e variação do lobo do Pleistoceno. J. Archaeol. Sci. 68 , 1-4 (2016).
- 4 Horard-Herbin, M.-P., Tresset, A. & Vigne, J.-D. Domesticação e usos do cão na Europa Ocidental desde o Paleolítico até a Idade do Ferro. Anim. Frente. 4 , 23-31 (2014).
- 5 Thalmann, O. et al. Os genomas mitocondriais completos dos canídeos antigos sugerem uma origem européia de cães domésticos. Science 342 , 871–874 (2013).
- 6 Savolainen, P., Zhang, Y.-P., Luo, J., Lundeberg, J. & Leitner, T. Evidência genética para uma origem do leste asiático de cães domésticos. Science 298 , 1610-1613 (2002).
- 7 Wang, G.-D. et al. Fora do sul da Ásia Oriental: a história natural dos cães domésticos em todo o mundo. Cell Res. 26 , 21-33 (2016).
- 8 Vonholdt, BM et al. As análises SNP e haplótipos em todo o genoma revelam uma rica história subjacente à domesticação de cães. Nature 464 , 898–902 (2010).
- 9 Shannon, LM et al. A estrutura genética em cães da vila revela uma origem de domesticação da Ásia Central. Proc. Natl Acad. Sci. USA 112 , 13639–13644 (2015).
- 10 Lüning, J. Erneute Gedanken zur Benennung der neolitischen Perioden. Germania 74 , 233-237 (1996).
- 11 Deguilloux, MF, Moquel, J., Pemonge, MH e Colombeau, G. O DNA antigo suporta a substituição de linhagem no pool genético de cães na Europa: uma visão sobre o sudeste neolítico da França. J. Archaeol. Sci. 36 , 513-519 (2009).
- 12 Frantz, LAF et al. Evidências genômicas e arqueológicas sugerem uma origem dupla de cães domésticos. Science 352 , 1228–1231 (2016).
- 13 Haak, W. et ai. A migração maciça das estepes foi uma fonte para as línguas indo-européias na Europa. Nature 522 , 207-211 (2015).
- 14 Seregély, T., Burgdorf, P., Gresik, G., Müller, MS e Wilk, A. «Tote Menschen und Tiere in Finteren Felsschächten…» - novos documentos documentais sobre métodos e métodos de avaliação unificada de Kirschbaumhöhle em Oberfranken. Praehistorische Zeitschrift 90 , 214-244 (2015).
- 15 O objetivo deste trabalho foi avaliar os efeitos de um teste de DNA em uma amostra de DNA de uma amostra de DNA de uma amostra de DNA. Bioinformatics 27 , 2153-2155 (2011).
- 16 Briggs, AW et al. Padrões de dano em seqüências genômicas de DNA de um neandertal. Proc. Natl Acad. Sci. USA 104 , 14616-14621 (2007).
- 17 Hofmanová, Z. et al. Os primeiros agricultores de toda a Europa descenderam diretamente dos egeus neolíticos. Proc. Natl Acad. Sci. USA 113 , 6886–6891 (2016).
- 18 Pilot, M. et al. Sobre a origem dos híbridos: história evolutiva de cães de criação livre na Eurásia. Proc. Biol. Sci. 282 , 20152189 (2015).
- 19 Duleba, A., Skonieczna, K., Bogdanowicz, W., Malyarchuk, B. & Grzybowski, T. Banco de dados completo do genoma mitocondrial e sistema de classificação padronizado para Canis lupus familiaris . Sci forense. Int. Genet. 19 , 123-129 (2015).
- 20 Larson, G. et ai. Repensando a domesticação de cães, integrando genética, arqueologia e biogeografia. Proc. Natl Acad. Sci. USA 109 , 8878-8883 (2012).
- 21 Rasmussen, M. et ai. O genoma de um ser humano do Pleistoceno Final de um cemitério de Clovis, no oeste de Montana. Nature 506 , 225-229 (2014).
- 22 Patterson, N. et ai. Mistura antiga na história humana. Genetics 192 , 1065-1093 (2012).
- 23 Lipson, M. et ai. Inferência eficiente no momento dos parâmetros da mistura e fontes de fluxo gênico. Mol. Biol. Evol. 30 , 1788-1802 (2013).
- 24 Bradburd, G., Ralph, PL & Coop, G. Uma estrutura espacial para entender a estrutura e mistura populacional. PLoS Genet. 12 , e1005703 (2016).
- 25 Gronau, I., Hubisz, MJ, Gulko, B., Danko, CG e Siepel, A. Inferência bayesiana da demografia humana antiga das sequências individuais do genoma. Nat. Genet. 43 , 1031-1034 (2011).
- 26 Freedman, AH et al. O sequenciamento do genoma destaca a dinâmica história inicial dos cães. PLoS Genet. 10 , e1004016 (2014).
- 27 Fan, Z. et al. Padrões mundiais de variação e mistura genômica em lobos cinzentos. Genome Res. 26 , 163-173 (2016).
- 28. Skoglund, P., Ersmark, E., Palkopoulou, E. e Dalén, L. O genoma do lobo antigo revela uma divergência precoce dos ancestrais de cães domésticos e mistura com raças de alta latitude. Curr. Biol. 25 , 1515-1519 (2015).
- 29 Song, S., Sliwerska, E., Emery, S. & Kidd, JM Modelando a história da separação da população humana usando genomas em fases. Genetics 205 , 385–395 (2017).
- 30 Axelsson, E. et ai. A assinatura genômica da domesticação de cães revela adaptação a uma dieta rica em amido. Nature 495 , 360-364 (2013).
- 31 Arendt, M., Fall, T., Lindblad-Toh, K. & Axelsson, E. A atividade da amilase está associada ao número de cópias AMY2B em cães: implicações para a domesticação, dieta e diabetes do cão. Anim. Genet. 45 , 716-722 (2014).
- 32. Reiter, T., Jagoda, E. & Capellini, TD Variação alimentar e evolução do número de cópias de genes entre raças de cães. PLoS ONE 11 , e0148899 (2016).
- 33 Anderson, TM et al. História molecular e evolutiva do melanismo em lobos cinzentos norte-americanos. Ciência 323 , 1339–1343 (2009).
- 34 Arendt, M., Cairns, KM, Ballard, JWO, Savolainen, P. & Axelsson, E. A adaptação da dieta em cães reflete a disseminação da agricultura pré-histórica. Heredity 117 , 301–306 (2016).
- 35 Allentoft, ME et al. Genômica populacional da Eurásia da Idade do Bronze. Nature 522 , 167-172 (2015).
- 36. Ramsey, CB Análise Bayesiana de datas de radiocarbono. Radiocarbon 51 , 337-360 (2009).
- 37. Reimer, PJ et al. Curvas de calibração por idade de radiocarbono IntCal13 e marine13 de 0 a 50.000 anos após a PA. Radiocarbono 55 , 1869-1887 (2013).
- 38. MacHugh, DE, Edwards, CJ, Bailey, JF, Bancroft, DR & Bradley, DG A extração e análise de DNA antigo de ossos e dentes: um levantamento das metodologias atuais. Anc. Biomol. 3 , 81-102 (2000).
- 39. Gamba, C. et al. Fluxo e estase do genoma em um transecto de cinco milênios da pré-história européia. Nat. Comum. 5 , 5257 (2014).
- 40. Bramanti, B. et al. Descontinuidade genética entre caçadores-coletores locais e os primeiros agricultores da Europa Central. Science 326 , 137-140 (2009).
- 41 Scheu, A. et al. A pré-história genética do gado domesticado, desde sua origem até a disseminação pela Europa. BMC Genet. 16 , 54 (2015).
- 42. Kircher, M. Análise de dados antigos de sequenciamento de DNA de alto rendimento. Métodos Mol. Biol. 840 , 197–228 (2012).
- 43 Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26 , 589–595 (2010).
- 44 DePristo, MA et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43 , 491–498 (2011).
- 45 Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30 , 2725–2729 (2013).
- 46. Paradis, E., Claude, J. & Strimmer, K. APE: análises de filogenética e evolução na linguagem R. Bioinformatics 20 , 289-290 (2004).
- 47 Patterson, N., Price, AL & Reich, D. Population structure and eigenanalysis. PLoS Genet. 2 , e090 (2006).
- 48. Alexander, DH, Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19 , 1655–1664 (2009).
- 49. Skotte, L., Korneliussen, TS & Albrechtsen, A. Estimating individual admixture proportions from next generation sequencing data. Genetics 195 , 693–702 (2013).
- 50. Jones, ER et al. Upper Palaeolithic genomes reveal deep roots of modern Eurasians. Nat. Comum. 6 , 8912 (2015).
- 51 Auton, A. et al. Genetic recombination is targeted towards gene promoter regions in dogs. PLoS Genet. 9 , e1003984 (2013).
- 52 Price, AL et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38 , 904–909 (2006).
- 53 Pavlidis, P. & Noble, WS Matrix2png: a utility for visualizing matrix data. Bioinformatics 19 , 295–296 (2003).
- 54 Sudmant, PH et al. Diversity of human copy number variation and multicopy genes. Science 330 , 641–646 (2010).
- 55 Alkan, C. et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat. Genet. 41 , 1061–1067 (2009).
- 56. Hach, F. et al. mrsFAST: a cache-oblivious algorithm for short-read mapping. Nat. Methods 7 , 576–577 (2010).
Agradecimentos
We thank Dan Bradley for his help obtaining the HXH specimen. We thank
Walter Eanes and Douglas Futuyma for their comments on the manuscript,
Dorina Twigg for the processing of canine copy number variation data,
Nick Patterson for providing advanced access to the latest version of
Admixtools, Vida for his thoughts, the NYGC for their assistance in the
sequencing, Valeria Mattiangeli for performing initial Miseq sequencing
on HXH, Christian Sell for his assistance with the raw data analysis
pipeline used for the CTC Miseq data, and the Musée zoologique de la
Ville de Strasbourg for hosting the team of archaezoology. The Kidd Lab
is supported by NIH Grant R01GM103961 and ALP is supported by
T32HG00040. AS and KD are supported by the EU: CodeX Project No: 295729.
Laura Botigué is supported by the Beatriu de Pinós Fellowship, from
Generalitat de Catalunya. SG was supported by a Boehringer Ingelheim
Fonds Travel award.
Informação sobre o autor
Afiliações
Contribuições
TS, AZ and RA provided the archaeological material. AS, SG, KD and MU performed the ancient DNA lab work and screening. LRB, SS, ALP, MO, AMT, DB, JMK and KRV performed the downstream bioinformatics and population genetic analysis. LRB, JB and KRV conceived the study. LRB and KRV wrote the paper.autor correspondente
Correspondence to Krishna R. Veeramah .Declarações de ética
Interesses competitivos
Os autores declaram não haver interesses financeiros concorrentes.Informação suplementar
Direitos e permissões
Open Access
This article is licensed under a Creative Commons Attribution 4.0
International License, which permits use, sharing, adaptation,
distribution and reproduction in any medium or format, as long as you
give appropriate credit to the original author(s) and the source,
provide a link to the Creative Commons license, and indicate if changes
were made. The images or other third party material in this article are
included in the article's Creative Commons license, unless indicated
otherwise in a credit line to the material. If material is not included
in the article's Creative Commons license and your intended use is not
permitted by statutory regulation or exceeds the permitted use, you will
need to obtain permission directly from the copyright holder. Para visualizar uma cópia desta licença, visite http://creativecommons.org/licenses/by/4.0/
Sobre este artigo

Citar este artigo
Botigué, L., Song, S., Scheu, A. et al. Ancient European dog genomes reveal continuity since the Early Neolithic. Nat Commun 8, 16082 (2017) doi:10.1038/ncomms16082
- Recebido
- Accepted
- Publicados
- DOI
assuntos
Leitura adicional
The curious case of the Mesolithic Iberian dogs: An archaeogenetic study
Journal of Archaeological Science (2019)Persistent Place-Making in Prehistory: the Creation, Maintenance, and Transformation of an Epipalaeolithic Landscape
Journal of Archaeological Method and Theory (2019)Origem e expansão recente de uma linhagem gammaretroviral endógena em canídeos domésticos e selvagens
Retrovirologia (2019)Rumo à identificação e papel das variações estruturais durante a domesticação de cães
National Science Review (2019)Exame genômico para seleção em galinhas da América do Sul revela região sob seleção associada à agressividade
Ciência Animal (2019)
Comentários
Ao enviar um comentário, você concorda em cumprir nossos Termos e Diretrizes da comunidade . Se você encontrar algo abusivo ou que não esteja em conformidade com nossos termos ou diretrizes, sinalize-o como impróprio.
Nenhum comentário:
Postar um comentário
Observação: somente um membro deste blog pode postar um comentário.